基因组设计与合成:从复写到理性设计
2021-05-22汪君仪武晓乐曹月阳李炳志
汪君仪,武晓乐,曹月阳,李炳志
(教育部合成生物学前沿科学中心,系统生物工程教育部重点实验室,天津大学化工学院,天津 300072)
快速发展的基因组测序技术推动了基因组信息的爆发式增长,正逐步加深人们对生命过程和调控的理解[1-3]。合成基因组学从复写已知天然基因组开始,逐步建立基因组合成技术,探索基因组设计原则,尝试以人工设计合成的基因组实现对生命活动的调控,深度拓展对生命过程的认知。
随着基因组测序技术的发展、DNA 合成与组装技术的进步、合成成本的降低,基因组的从头合成与重新设计日益受到关注。2002 年脊髓灰质炎病毒的基因组成为世界上第一个人工合成的基因组[4],2003 年φX174 噬菌体的基因组也被成功合成[5],2020年新冠肺炎病COVID-19基因组也被迅速从头化学合成并组装出有活性的毒株[6]。随着DNA 大片段合成组装技术的发展,2008 年原核生物生殖支原体的基因组被成功从头合成[7]。2010 年Venter 团队实现了利用人工合成的蕈状支原体基因组调控生命活动的目标[8],证实了化学合成基因组的生命活性。随着包括染色体构建与转移等基因组合成相关技术的发展[9-14],合成基因组学的研究可以在更多的物种范围和更大的设计尺度上探究基因组的基本科学问题,例如,在大肠杆菌中尝试进行基因组密码子的删减研究[15-18],在支原体中进行大规模基因组简化的研究[19],在酿酒酵母中进行基因组稳定性和柔性的研究[20-28],等等。真核单细胞模式生物酿酒酵母基因组的设计与合成成为科学家的又一个重大目标。
在合成基因组学发展的初期,目标主要是利用化学合成的手段成功复写野生型基因组,关键在于建立基因组合成的技术方法。近年来合成基因组学不断地发展,国内外也形成了不少相关综述[29-31],研究者逐渐开始利用合成基因组学探索生物学基本问题和原理,这就需通过对基因组进行不同的设计并以基因组合成的方式验证设计的可行性。本文作者将从人工基因组的密码子改变、人工标签、人工位点、基因组简化等几种重要设计角度,介绍基因组合成的相关研究进展。
1 人工基因组的密码子改变
1.1 密码子丰度的转变
密码子丰度是表征某一个密码子在全基因组编码中使用频率的指标。遗传密码具有简并性,这导致了对应同一种氨基酸会存在多个不同的密码子(同义密码子),同时,在不同生物间,甚至在同一生物不同蛋白间,编码相同氨基酸的同义密码子的使用频率不同,这就导致了密码子丰度的差异,通常也称之为密码子使用的偏好性[32]。为了让异源基因更好地在宿主中表达,根据宿主细胞密码子的偏好性对异源基因进行优化已经成为基因组设计中一种成熟的技术手段,已有多种密码子优化软件被开发,如Codon Optimization OnLine[33]。研究表明在酿酒酵母体内将同义密码子中相同的密码子排列在一起,可以使tRNA 分子循环利用从而加快翻译速度[34]。密码子分布与基因的GC 含量密切相关,连续同义密码子的选择具有相关性,倾向于选择使用相同tRNA的密码子。
Venter 团队在蕈状支原体基因组(JCVIsyn3.0)的合成过程中,对密码子丰度转变的原则进行了一定程度的探究,野生型蕈状支原体基因组中腺嘌呤(A)和胸嘧啶(T)的含量极高,TGA 仅作为编码色氨酸的密码子而并非终止密码子,并且偶尔会使用非标准的起始密码子。该研究将包含三个必需基因的5000 bp 区域中密码子的编码原则进行了三种不同方式的修改:①将编码色氨酸的密码子由TGA 更改为TGG,采用非标准的起始密码子;②根据大肠杆菌密码子的偏好性对该区域的密码子丰度进行修改,色氨酸仍由TGA 编码;③根据大肠杆菌密码子的偏好性对该区域的密码子丰度进行修改,使用标准的起始密码子,色氨酸由TGG 编码。实验结果表明这些密码子更改基本不影响细胞生长,但若在基因组设计中想要大规模改变密码子的丰度可能需要伴随tRNA水平的修饰以保证翻译能够正常进行[19]。
1.2 密码子删除
自然界中大多数生物均使用64 个遗传密码子编码20 种标准氨基酸,密码子具有通用性,即不同的生物使用一套高度相似的密码子表。通过在全基因组范围内进行同义密码子的替换并消除其对应的tRNA,使其无法翻译所删除的密码子,则无法正确翻译来自病毒、质粒和其他细胞获得的DNA,从而使重新编码的生物具备遗传隔离能力。并且我们可以利用所删除的密码子来编码非标准氨基酸,合成具备新化学性质的蛋白,也可利用非标准氨基酸来建立生物防逃逸体系。
为了进一步探索密码子的使用规则及遗传信息的保存、编码、交换原理,2013 年Church 团队提出创建使用63 个密码子的生物。在大肠杆菌MG1655 中采取基因编辑的方式利用同义密码子UAA 替换了所有UAG 终止密码子,并通过删除RF1(释放因子)消除了在密码子UAG处终止翻译的功能,从而可以将UAG 用于特异性地编码非标准氨基酸[15]。在成功完成单密码子替换工作的基础上,进一步开始探索在全基因组范围内进行多个密码子替换的可行性。2016年,在大肠杆菌全基因组范围内使用同义密码子系统地替换了7 个密码子(包含6个有义密码子以及UAG终止密码子),使大肠杆菌的遗传密码子从64 个减少到57 个,共涉及到62 214 个同义密码子的替换,但最终仅包含57个密码子的大肠杆菌无法正常存活,至今也未能成功构建。完成替换后将重新编码的基因组解析为87 个约50 kb 的片段,并对其中55 个片段(包含2229 个基因)进行基因功能的验证,其中仅有13 个重新编码的必需基因无法支持细胞生存,99.5%重新编码的基因无需任何优化即可替代野生型基因的功能[16],这在一定程度上表明了从根本上改变遗传密码和大规模合成基因组重构的可行性。
利用基因组的重新编码以去除部分有义密码子,可以促进具备非天然氨基酸编码功能的细胞的合成。2016 年Chin 团队提出了利用大于100 kb的DNA 合成片段迭代替换相应野生型片段的基因组组装技术,在进行多次迭代后可以完成整个大肠杆菌基因组的替换,该方法为全基因组同义密码子的替换提供了基础。此外该研究尝试使用了8种不同的同义密码子替换方案,并成功地在富含必需基因的操纵子中去除了373 个有义密码子[17]。2019年,在此研究基础上,利用基因组全合成的方式完成了大肠杆菌全基因组范围内2 个有义密码子和1个终止密码子的替换,并且仅含有61种密码子的大肠杆菌可以维持正常的生长,该研究重新编码了18 214 个密码子,成功创建使用61 个密码子的生物,并删除了其对应的tRNA[18](图1)。
2 人工标签
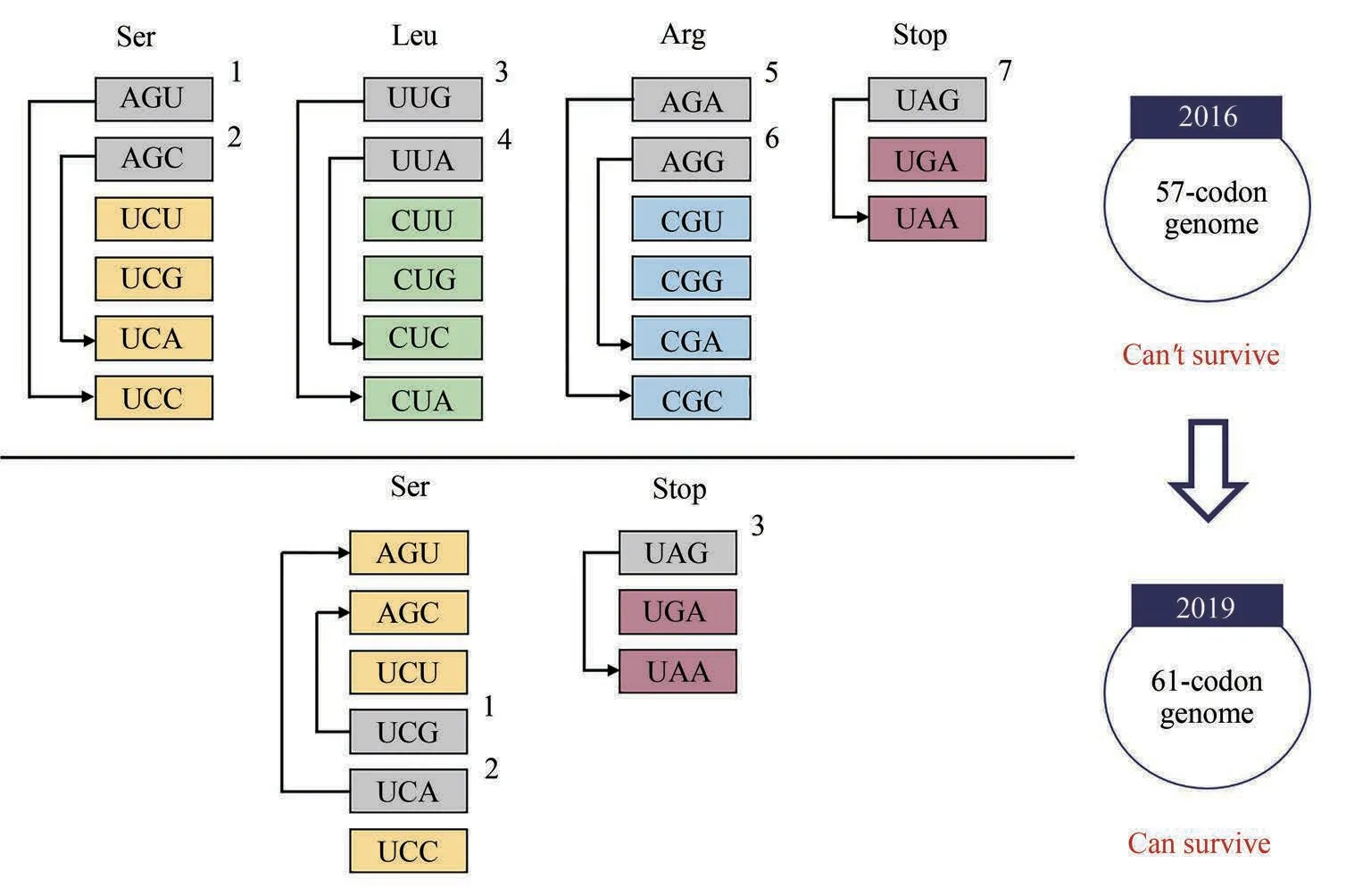
图1 密码子删除相关研究进展[16,18](2016年Church团队在大肠杆菌全基因组范围内使用同义密码子系统地替换了7个密码子,90%以上的基因仍具有功能[16] ;2019年Chin团队重新编码了大肠杆菌的18 214个密码子,创建了使用61个密码子的生物[18])Fig.1 Research progress on codons deletion[16,18](In 2016,seven codons were replaced with synonymous alternatives the entire genome of E.coli by Church's group,more than 90%of the genes retained functionality[16];In 2019,Chin's group recoded 18 214 codons to create a strain with a 61-codon genome [18])
2010 年,在合成蕈状支原体基因组研究中,为了区分合成基因组和天然基因组,在不干扰细胞活力的前提下在合成型基因组中引入了四个“水印”序列[8]。在Sc2.0 项目中,利用同义密码子替换,在合成型基因组中引入了大量的PCR 标签,用于区分合成型和野生型序列[20]。除此之外,Sc2.0 的设计还包括内含子及重复序列的删除、tRNA 的移位等[35]。在synⅩ染色体的合成过程中,Wu 等[25]开 发 了 一 种 名 为 混 菌PCR 标 签 定 位(pooled PCRTag mapping, PoPM)的高通量定位策略,用于高效识别合成型染色体中导致生长缺陷的位点。该方法以生长缺陷和长势正常两个混菌库提取的基因组作为模板,以PCRtag 为引物进行PCR 反应,通过结果分析正常菌株表型库中缺失的合成型区域和生长缺陷菌株库中缺失的野生型区域,快速定位导致缺陷的基因组位点。
除此之外,利用PCRtag 可以便捷地分析合成型基因组重排前后变化的基因组位点,是一种有效的快速缩小潜在目标基因位点所在区域的方法。Luo 等[36]利用基因组重排对酵母菌株乙醇的耐受性机制进行了探索,将重排后的菌株分为耐受乙醇组和不耐受乙醇组,利用合成型基因组上的PCRtag 对这两组菌株进行PCR 分析,找到了影响乙醇耐受性的基因位点。在利用含有合成型染色体菌株的基因组重排研究中,利用合成型染色体中非必需基因上包含的96 个PCRtag 对7 株重排后获得的菌株进行分析,发现其中4株菌株都包含一段长度约为11 448 bp 区域的缺失,在这段区域内进一步进行基因的删除分析,获得了碱性耐受相关基因的位点[37]。
3 人工位点
染色体的结构以及基因的排列顺序都会影响基因的表达,利用合成型基因组重排快速探究菌株更优的生理表型受到广泛的关注。Sc2.0 项目中合成型酵母染色体设计的一大亮点是在每个非必需基因的终止密码子后均插入了一段反向对称的人工位点-loxPsym 序列[35],两个loxPsym 位点之间的序列在Cre重组酶的作用下可随机发生删除和反转[38],从而在合成型酵母体内形成了可快速进行基因组重排(缺失、重复、倒置、易位)的SCRaMbLE (Synthetic Chromosome Rearrangement and Modification by LoxPsym-mediated Evolution)系统。为了证明SCRaMbLE 系统在探索基因组多样性方面的能力,利用合成染色体臂synIXR(包含43 个loxPsym 位点)进行基因组重排,其中64个SCRaMbLE 菌株深度测序结果显示,重排后除了可以获得基因组缺失、重复、倒位的菌株还可以获得发生大量复杂重排的菌株,并且这些重排均发生在所设计的loxPsym 位点之间[39]。利用SCRaMbLE 系统可以快速地进行大规模的基因组重排(包含染色体内及染色体间的重排),在选择性条件下可快速有效地进行基因组的定向进化。SCRaMbLE 系统不断在应用中得到改进,逐步使SCRaMbLE 系统成为优化宿主、提高产量以及增强菌株耐受性的有效手段。
3.1 SCRaMbLE系统的改进
最初设计的SCRaMbLE 系统是以雌激素诱导为重排开关,但在缺乏雌激素诱导的情况下仍然可以观察到染色体重排引起的合成型酵母的生长缺陷,这表明了单开关控制下的SCRaMbLE 系统存在开关的泄漏[39]。Jia 等[40]将半乳糖驱动的Cre酶诱导表达系统引入最初的设计体系,通过构建与门开关实现了对SCRaMbLE 系统的精准控制。与门开关结合了转录控制以及细胞定位控制,使SCRaMbLE 系统只有在同时具备半乳糖和雌激素的情况下才可开启。在此基础上,进一步开发了多重SCRaMbLE 迭代循环(Multiplex SCRaMbLE Iterative Cycling,MuSIC)技术以产生更多的合成基因组重排变体。Cre 酶表达的泄漏还会导致重排结束后基因组的不稳定,为了维持重排后基因组的稳定性,Lin 等[41]在酿酒酵母中导入与Cre重组系统正交的Vika 重组系统,利用Vika-vox 系统在重排结束后彻底删除Cre基因以维持重排后菌株的稳定性。
尽管雌激素是一种强力诱导剂,但它具有的活性在浓度较高的情况下可能对人体有毒害。Hochrein 等[42]开发了一种光控拆分的Cre重组系统(light-controlled induction system, L-SCRaMbLE),该系统中Cre 酶被分裂为N 端和C 端,并分别融合到可以光学诱导二聚化的两个蛋白上,在红光介导的二聚作用下重构Cre 重组酶。L-SCRaMbLE 操作更加简便,并且可以通过调节感光体发色团的浓度、光照射时间以及光照强度来微调重组效率。虽然L-SCRaMbLE 系统总体重组效率较低,但较低的重组效率更有利于产生重组多样性[42]。
利用SCRaMbLE 系统筛选对某些胁迫因素具有更高耐受性的菌株时,发现很多菌株耐受性的提高并不是染色体重排导致。为了有效鉴定细胞是否发生染色体重排,Luo 等[36]基于营养缺陷型标签URA3 和LEU2 的交替切换设计了ReSCuES(reporter of SCRaMbLEd cells using efficient selection)系统。未发生SCRaMbLE 时菌株能在缺乏尿嘧啶的培养基中存活,只有发生染色体重排的菌株才能在缺乏亮氨酸的培养基中存活,该系统可快速鉴别发生重排的菌株。
单倍体合成型酵母在发生SCRaMbLE 时,必需基因的缺失使基因组重排存在高致死率的问题,为此Shen等[43]开发了杂合二倍体的SCRaMbLE技术。该研究将单倍体合成型Sc2.0 菌株与单倍体天然亲本菌株交配产生杂合二倍体菌株,以此杂合二倍体菌株进行SCRaMbLE,并且该技术也可以在种间杂交中应用。利用二倍体菌株进行SCRaMbLE 降低重排的致死率,通过环境选择可更快更多地产生新的表型。
为了使SCRaMbLE 系统更加可控和便捷,进一步开发了自上而下和自下而上的体外SCRaMbLE系统,该系统由试管中的Cre 重组酶驱动,同时存在含有多个loxPsym位点DNA片段,通过相关转录单元的重排来优化生物合成的通路[44]。Liu 等[45]提出了一种基于重组酶的方法(recombinase-based combinatorial method, SCRaMbLE-in),包含一个体外重组酶系统,可以在途径水平上快速实现基因表达的多样化,同时开启体内SCRaMbLE 系统将体外组装的路径整合到合成型酵母基因组中,并在宿主细胞中进行大量的基因组重排,以同时达到外源途径优化和底盘菌株优化的效果。
3.2 SCRaMbLE系统的应用
合成型酵母染色体的重排是优化宿主、提高产量以及增强菌株耐受性的有效手段。研究人员利用含有合成型染色体的酿酒酵母进行SCRaMbLE,得到了可以利用木糖的宿主细胞,提高了酿酒酵母中紫色杆菌素、脱氧紫色杆菌素前体、青霉素、β-胡萝卜素、类胡萝卜素等次级代谢产物产量[40,46-47]。并且通过重排产生了乙醇耐受性增强、耐碱性增强的菌株,同时对酿酒酵母的耐乙醇机制及耐碱机制进行了更加深入的探究[36-37]。此外利用SCRaMbLE 系统进行基因组规模的基因敲除是研究基因间相互作用的有效途径之一,可以利用SCRaMbLE 系统随机产生缺失突变体,以此来探索基因合成致死的相互作用[48]。随着SCRaMbLE系统应用的逐渐成熟,研究人员开发了一种基于此系统的基因组简化方法(SCRaMbLE-based genome compaction,SGC),并通过迭代SGC 实现了合成型染色体臂(synXIIL)中39 个非必需基因的删除[49]。
4 基因组简化
在自然界中大多数生物为了能够适应多种环境,体内含有很多与细胞基本生长无关的基因,简化基因组的研究可以促进基因功能的解析,减少基因组中冗余的基因,增加细胞对能量的利用效率,增强细胞的可控性,有助于生物遗传调控的研究,为合成基因组的设计提供基础。
近年来,基因组简化研究也取得了快速发展(图2)。利用Tn5 转座子的衍生物将E.coli K-12 的基因组减小了200 kb[50],在粟酒裂殖酵母体内敲除了冗余的223 个基因,将基因组减小了657.3 kb[50]。使用改良的转座子诱变法,经过不断地设计、合成、测试,确定了维持细胞生长的必需及准必需基因,构建了简化的蕈状支原体JCVIsyn3.0 菌株,该基因组(531 kb)大小仅有原基因组JCVI-syn1.0 (1079 kb)的1 2 左右,比自然界中已知可独立存活的最小基因组M. genitalium(580 kb)还要小[19]。该简化过程中删除了428 个基因,但仍有149个功能未知的基因,这表明还存在着某些维持生命所必需的功能是未知的,JCVIsyn3.0 可作为进一步基因组功能研究以及全基因组设计探索的平台[19]。
5 展 望
合成基因组学研究发展迅速,从病毒基因组和原核生物基因组的全合成,已经快速发展到真核生物基因组的合成阶段,酿酒酵母6条染色体已经完成人工设计合成。现在已经提出了基因组编写计划[52],将有更多的多细胞生物基因组进入设计合成研究的计划。但目前合成基因组学的发展仍处于起步阶段,还面临着很多技术的挑战。
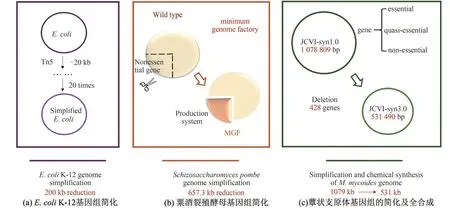
图2 基因组简化的研究进展[19,50-51][(a)利用Tn5转座子的衍生物将大肠杆菌K-12的基因组缩减200 kb[50];(b)利用LATOUR删除法将粟酒裂殖酵母基因组缩减657.3 kb[51];(c)利用全基因组设计和化学合成的方法简化支原体JCVI-syn1.0(1079 kb),通过设计、合成、测试三个步骤的循环,保留了必需基因和准必需基因,得到支原体JCVI-syn3.0(531 kb,473个基因)[19]]Fig.2 Advances in genome simplification[19,50-51][(a)Simplification of the E.coli K-12 genome.The deletion procedure has reduced the genome at an average of 200 kb by using specialized transpo‐sons (Tn5 derivatives) to create deletions in the E. coli K-12 chromosome[50]. (b) Simplification of the Schizosaccharomyces pombe genome. Re‐searchers have reduced the genome of S. pombe by 657.3 kb using a large-scale gene deletion method called LATOUR[51]. (c) Simplification and chemical synthesis of M. mycoides genome. Using whole-genome design and complete chemical synthesis, researchers have minimized the 1079-kilobase pair synthetic genome of M.mycoides JCVI-syn1.0.Three cycles of design,synthesis, and testing,with the retention of essential and quasiessential genes,produced JCVI-syn3.0(531 kilobase pairs,473 genes)[19]]
基因组的理性设计需要借助更多的技术手段,例如利用计算机辅助设计(computer-aided design , CAD)进行的大规模基因组设计以及利用计算机软件进行基因组的可视化和质量控制等。目前使用的合成基因组CAD 软件虽然可以进行大规模的染色体水平DNA 序列设计,但设计后基因组的功能验证仍然只能通过实验来完成。开发根据基因组设计预测生物生存能力及表型的软件还面临着巨大的挑战。伴随着人工智能的发展,大量生物信息的整合,将来可以通过机器学习的方法来促进人工基因组表型的预测。然而,目前数据不兼容及缺乏足够的描述性元数据的问题还有待解决[29]。我们对基因组功能的了解逐渐深入,可以获得的生物基本数据快速增多,这将可以帮助我们进一步加大设计尺度,探究基因组设计的边界。
Sc2.0 项目中提出的SCRaMbLE 系统可在短时间实现基因组大规模的重排, SCRaMbLE 系统的诱导方式、重排效率、应用对象等方面也在不断地改进,但目前SCRaMbLE 系统大多只应用于含有单条或两条合成型染色体的菌株中。在酵母全基因组合成完成后,全基因组重排系统将会产生更多更复杂的染色体内和染色体间的基因组重排。另一方面,目前SCRaMBLE 系统完全依赖于Creloxp 位点特异性重组系统,还可以将与Cre 酶正交的重组酶系统引入已完成基因分区的细胞内,构建可诱导的基因组模块化重排系统。越来越多的酪氨酸重组酶系统被发现,从最初的Cre/Loxp 和FLP/FRT系统到新发现的Vika/vox、Dre/rox、VCre/Vloxp、SCre/Sloxp 等系统均被证明可在异源宿主中发挥重组作用[53],这些重组酶均可用于尝试构建正交化的重组系统。利用基因组模块化重排系统有望解析不同的基因组合与生物性能的关联性。
目前针对基因组简化的方法主要是从理性简化的角度,对已知的冗余基因、重复序列、高度可变区、转座子以及内含子等进行删除,通过不断“设计-合成-测试”的循环来实现基因组的简化,工作量较大。利用SCRaMBLE系统进行多轮随机删除,还可在保证菌株适应度的基础上实现基因组的持续简化,进一步探究基因组简化的规律。
