亚丁牦牛和拉日马牦牛遗传多样性及遗传结构分析
2021-05-21纪会官久强王会周建旭阿农呷何宗伟樊珍详邱龙康曹诗晓安添午柏琴钟金城罗晓林
纪会 ,官久强 ,王会 ,周建旭 ,阿农呷 ,何宗伟 ,樊珍详 ,邱龙康 ,曹诗晓 ,安添午 ,柏琴 ,钟金城 *,罗晓林 *
(1. 四川省草原科学研究院,四川 成都 611731;2. 西南民族大学,四川 成都 610041;3. 小金县科学技术和农业畜牧水务局,四川 阿坝州 624200;4.甘孜藏族自治州畜牧站,四川 甘孜州 626099;5. 稻城县农牧农村和科技局畜牧站,四川 甘孜州 627750;6. 新龙县农牧农村和科技局畜牧站,四川 甘孜州 626800)
牦牛(Bos grunniens)是分布于以青藏高原为中心,及其毗邻的高山、亚高山地区的特有珍稀牛种[1]。青藏高原地域环境复杂,不同区域的牦牛在生态适应性方面存在明显差异,表现为体型外貌和生产性能的差异,形成了各具特色的地域类型和亚型[2]。家牦牛约在7300 年前由野牦牛驯化而来[3],中国有18 个地方品种(遗传资源)和2 个培育品种,是世界上拥有牦牛数量和品种类群最多的国家。然而由于长期过度选育和纯繁,导致现有家养牦牛出现品种流失的现象,尤其是与其抗病性、肉质等相关性状的退化已相当严重,极大地影响了青藏高原及周边地区农牧业生产的稳定性[4]。因此,加强地方牦牛群体遗传多样性研究,挖掘新种质资源,对于遗传资源保护、新品种培育和牦牛生产性能提高等都具有积极的理论和现实意义。
亚丁牦牛和拉日马牦牛是肉乳兼用型优良地方牦牛资源。亚丁牦牛分布于四川省甘孜藏族自治州稻城县,经产母牛 153 d 平均挤奶量 434 kg,产奶量居中国牦牛之最[5−6],2005 年亚丁牦牛存栏量约 7.5 万头,2015 年降至4.6 万头,由于饲草料缺乏,背山封闭,近亲交配严重,繁殖性能差,其群体有退化趋势。拉日马牦牛中心产区位于四川省甘孜藏族自治州新龙县拉日马镇,2016 年存栏量约8.7 万头,具有净肉率高的特性,公牛净肉率达45.7%[7]。然而,关于两个群体分子遗传背景的研究尚未开展。
限制性内切位点相关的DNA 标记(restriction site-association DNA,RAD)简化基因组测序,即利用限制性酶切法获得目的基因组部分片段进行高通量测序,获得可代表目的基因组信息的高密度SNPs 分子标记,具有简便、有效及低成本等特点,已广泛用于群体遗传多样性研究,如探究孟加拉虎出现白色条纹的遗传机理[8],对鹿苑鸡不同保种群保种现状的评估[9]及中国家兔地方品种的遗传地位研究[10]。本研究利用RAD 简化基因组测序对9个地方牦牛群体进行全基因组限制性酶切检测,以期在DNA 分子水平分析亚丁牦牛和拉日马牦牛的遗传多样性,探明各群体间的进化关系,为今后进一步开展两种牦牛遗传资源的保护、遗传改良和育种提供理论依据。
1 材料与方法
1.1 试验动物和方法
以亚丁牦牛、拉日马牦牛、九龙牦牛、麦洼牦牛、金川牦牛、昌台牦牛、中甸牦牛、玉树牦牛、类乌齐牦牛为试验动物。每个群体随机选择9~11 头,采集类乌齐牦牛的组织样本以及其他8 个牦牛群体的血液样本(表1)。具体操作:用一次性注射器从牦牛颈外静脉采集5 mL 血液于EDTA 抗凝管,放入冰盒中带回,放置−80 ℃冰箱中保存;对类乌齐牦牛进行屠宰后采集组织样品,经DEPC(diethyl pyrocarbonate,焦碳酸二乙酯)冲洗后,锡箔纸包装迅速置于液氮保存,带回实验室备用。牦牛群体地理分布情况见图1。
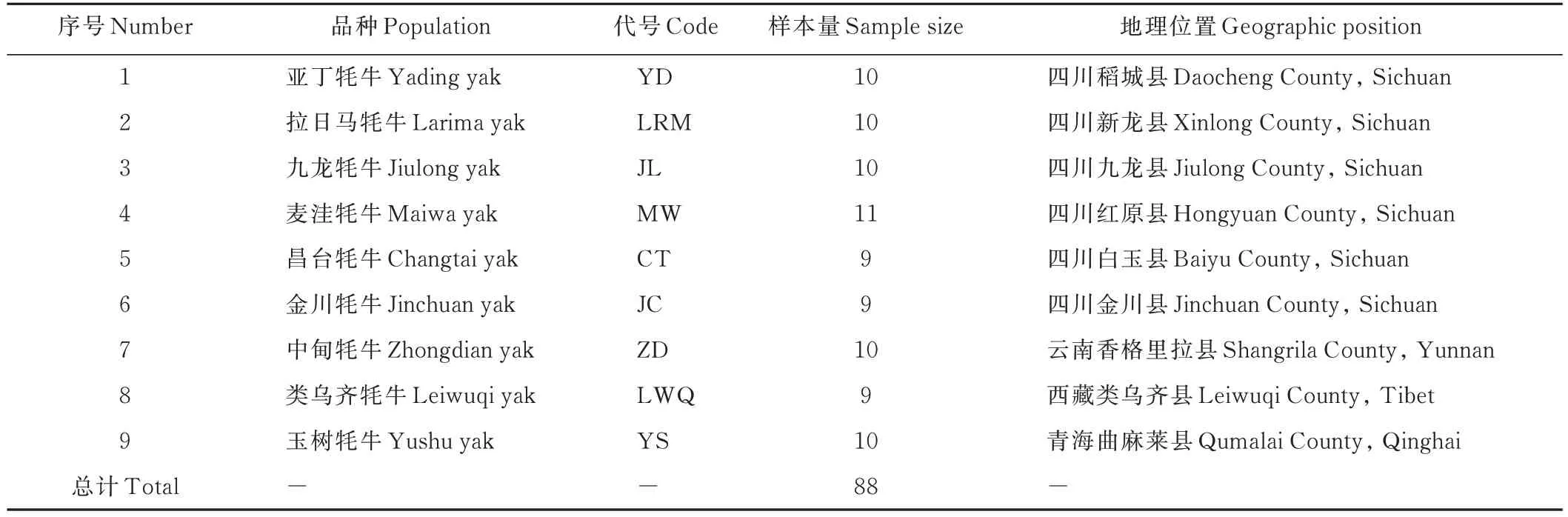
表1 牦牛样本来源及样本量Table 1 Source and number of yak sample
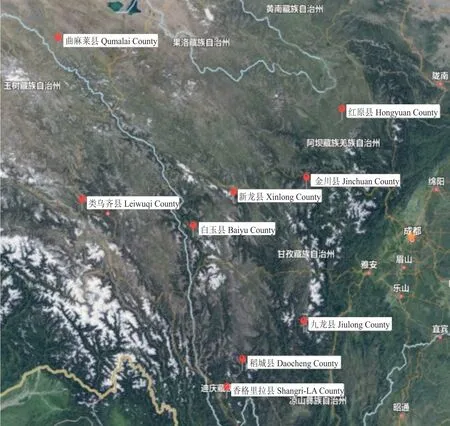
图1 牦牛样本的地理分布Fig.1 Geographical distribution of yak sample
1.2 基因组电子酶切评估
建库测序前,为检测酶切效率和对所有标记进行分析,按照PstI 酶(CTGCA,G)切位点对参考基因组(GCF_000298355.1)进行模拟酶切(电子酶切),统计获得限制性酶切片段的数量及覆盖率。共获得2959206 条限制性酶切片段(restriction fragment,RF),基因组覆盖率为41.89%,其中有效限制性酶切片段(effective RF:长度大于300 bp)共1558672 条。有效限制性酶切片段在基因组中分布均匀,PstI 酶对牦牛参考基因组的酶切效果较好,可用于建库测序(图2)。
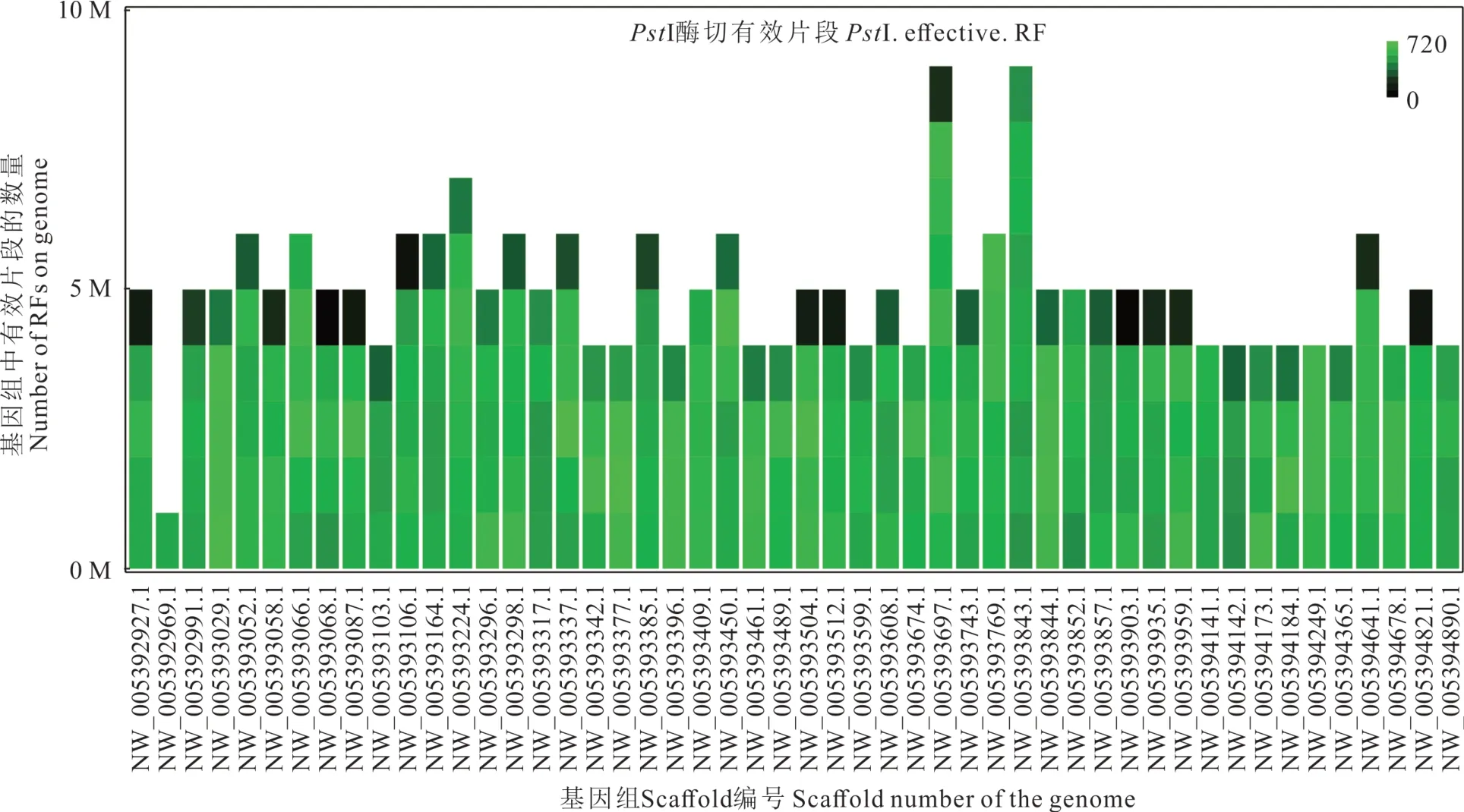
图2 基因组中有效限制性酶切片段分布图Fig.2 Map of effective restriction fragments(effective-RF)in the genome
1.3 全基因组限制性酶切
采用血液基因组DNA 试剂盒(北京天根)提取血样DNA,CTAB(cetyltrimethylammonium bromide,十六烷基三甲基溴化铵)法提取组织样DNA。采用PstI 酶对全基因组进行酶切,88 个样本共构建88 个简化基因组测序(restriction site-association DNA,RAD)文库,在HiSeq3000 测序平台上用150 bp 双端序列进行RAD 测序。
1.4 数据处理
测序数据的质量控制和原始数据过滤:通过分析碱基的质量值和组成评估数据的质量。对原始序列(raw reads)中低质量的reads 进行过滤,获得高质量的净数据(clean reads),用于后续的信息分析。过滤条件:去除带接头的reads;去除含未知碱基比例大于10%的reads;去除低质量reads(质量值Q≤20 的碱基数占整条reads 的50% 以上)[11]。
单核苷酸多态性(single nucleotide polymorphisms,SNP)的识别与过滤:使用比对软件 bwa[12](0.7.12)采用mem 算法将过滤后的clean reads 比对到参考基因组上,比对参数为-k32-M;比对结果使用picard(1.129)软件标记 duplicates reads(PCR 过量扩增所形成的 reads)。使用软件 GATK[13]检测群体 SNP/indel,参数:-filter“QD<2.0”,得到可信度较高的SNP/indel 数据集。Indel(insertion-deletion)全称为插入或缺失突变,指在DNA 或RNA水平上,发生于个体间的单核苷酸突变位点。为提高分析准确性,对原始SNP 进行过滤,过滤条件为:缺失率(missing rate)不超过20%,第二等位基因频率(minor allele frequency,MAF)不小于0。
1.5 群体遗传多样性分析
杂合度统计:杂合度指同源染色体上的一个位点存在不同等位基因的状态,用于衡量群体中的遗传变异程度[14]。应用 Stacks populations v 2.41 软件[15],其参数设置如下:最小等位基因频率(MAF)>0.05;标签最低覆盖倍数>3×;在每个等位基因位点处,每个品系至少75%的个体存在序列信息,至少3 个品系存在有效信息[16]。杂合度取值在0~1 之间,杂合度越高,表明群体内遗传多样性系数越高。
连锁不平衡(linkage disequilibrium,LD)分析:连锁不平衡指群体内不同座位等位基因之间的关联。采用PopLDdecay 3.31[17]软件,通过计算一定序列范围内两两位点间的LD 系数(r2),来估算LD 衰减的趋势。r2代表两位点的相关性,它在0<r2<1 的区间内变化,如果r2=0,表示两位点发生重组的可能性是100%,如果r2=1,则表示位点完全连锁。通常位点间的关联性会随着基因组上位点间距离的增加而不断下降,称为LD 衰减速度。使用LD 衰减距离来描述LD 衰减速度的快慢,衰减距离越长,其衰减速度越慢,群内等位基因间连锁程度越强。
1.6 群体间遗传关系分析
成对遗传分化值(Pairwise Fst)[18]:分析不同品种间的遗传距离。应用Stacks populations v 2.41 软件,参数设置与杂合度统计一致。根据每个等位基因位点SNP 信息,进行双向选择家系内Pairwise Fst 统计,并对各Fst 进行Fisher 精确检验,使用P 值对Fst 值进行校正(Corrected Fst),进而根据高斯核函数,计算Smoothed pairwise Fst,利用 40 kb-Slid Windows 对全基因组上的 Fst 进行平均值统计。采用 Wright[19]理论,如群体 Fst 值为 0.00~0.05,说明群体间不存在分化;Fst 值为0.05~0.15,说明群体间处于中度分化;Fst 值为0.15~0.25,则说明群体间分化程度较大;Fst 值大于0.25,说明群体间存在极大分化。
主成分分析(principal component analysis,PCA):使用软件GCTA[20]从中获得各个样本的主成分值。根据各个样本在第一主成分(PC1)、第二主成分(PC2)、第三主成分(PC3)3 个综合指标中的数值大小,使用R 语言绘制PCA 散点图。散点图中每一个位点代表一个样本,图中两个样本距离越远,则说明两个样本遗传背景差异越大[21],遗传背景相似的个体则会在图中聚为一类。
系统进化树构建:基于群体SNP 数据运用MEGA 4.0[22]软件计算样品间距离,得到个体间的序列差异度矩阵,然后使用IQ-Tree 软件,最大似然法构建系统进化树(maximum likelihood tree,ML-tree)。
1.7 群体结构分析
应用Structure[23]软件进行群体结构分析,用于推算测序样本分属几个群体,群体间基因交流程度以及每个样本的混血程度。基于多位点基因型将遗传相似的个体聚类。先预设88 个样本由若干亚群K(K=1~n)构成,根据每个SNP 位点的基因型采用隐马科夫−蒙特卡罗链模型算法(markov-chain monte carlo,MCMC)[24],在每个群体均最大程度符合哈代温伯格平衡检验前提下,输出88 个样本被分为K个亚群时的最佳分类状态,同时每一个K值均会对应产生一个似然值(likelihood),最后对似然值取自然对数后输出Inlikelihood,Inlikelihood 越大,说明K值越接近于真实情况,一般Inlikelihood 值先随着K值增大而不断升高,之后进入平台期,最佳K值即为ΔK值最大时。
2 结果与分析
2.1 牦牛基因组的SNP
对RAD 简化基因组文库进行Illumina Hiseq 3000 双端高通量测序,88 个牦牛样本测序获得4.2 G reads 原始序列数据,经过滤后有效数据量为3.9 G。各群体原始数据有效率在89.82%~95.54%,测序质量数值大于20、30 的碱基占总体碱基的百分比为Q20≥96.93%、Q30≥91.47%,GC 含量在48.35%~49.07%,说明碱基组成平衡,测序质量较好,数据可靠。将过滤后的高质量reads 与参考基因组比对,比对率均在98.00%以上。平均测序深度6.8×,88 个样本基因组覆盖率在30.99%~38.70%,与电子酶切参考基因组预测的覆盖率相近,预期率达92.48%,说明RAD 简化测序预测牦牛基因组效果较好(表2)。
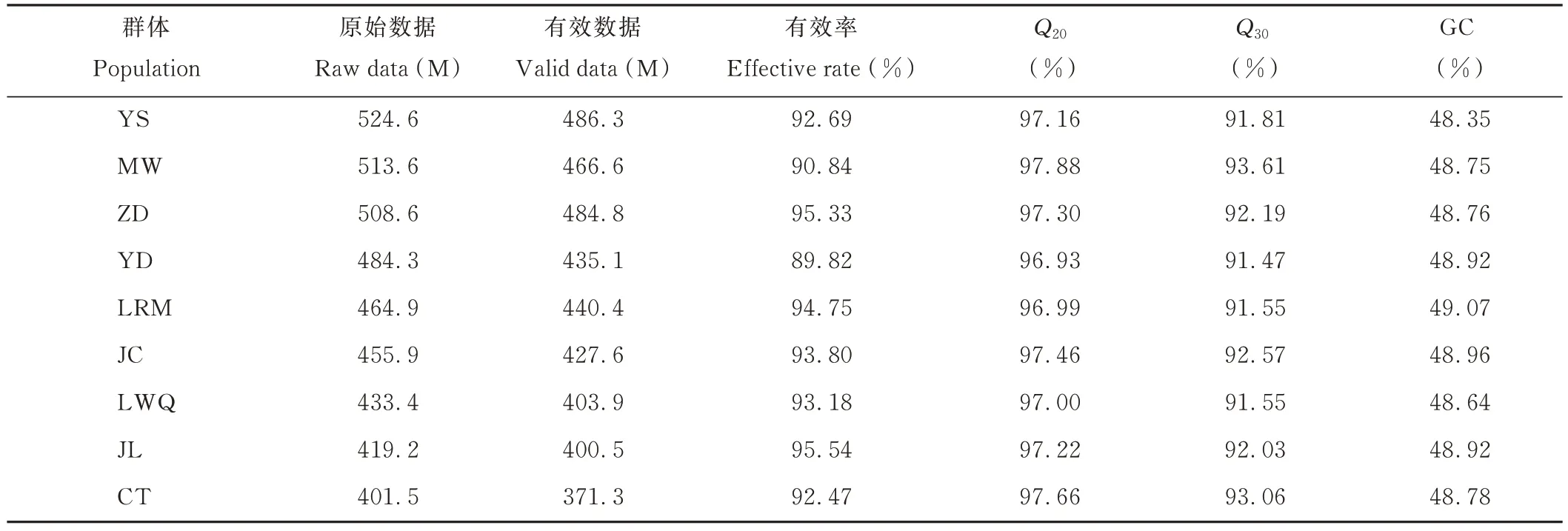
表2 测序数据质量汇总情况Table 2 Summary of sequencing data quality
所有样本共检测到20413266 个SNP 位点,亚丁牦牛SNP 数量最多,为7596791,昌台牦牛SNP 数量最少,为5472579,拉日马牦牛SNP 数量为6163048,不同群体SNP 信息见图3。另检测到插入变异位点(insertion)有 362758 个 ,缺 失 变 异 位 点(deletion)487510 个,插入和缺失位点长度集中在1~10 bp,其中1~3 bp 最多(图4)。
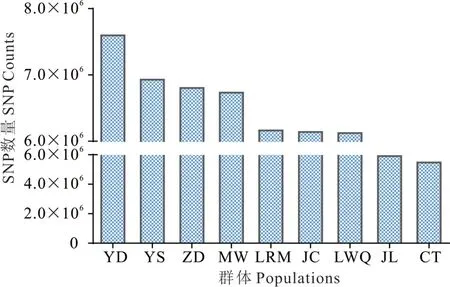
图 3 9 个群体 SNP 数量Fig.3 Number of SNP in 9 populations
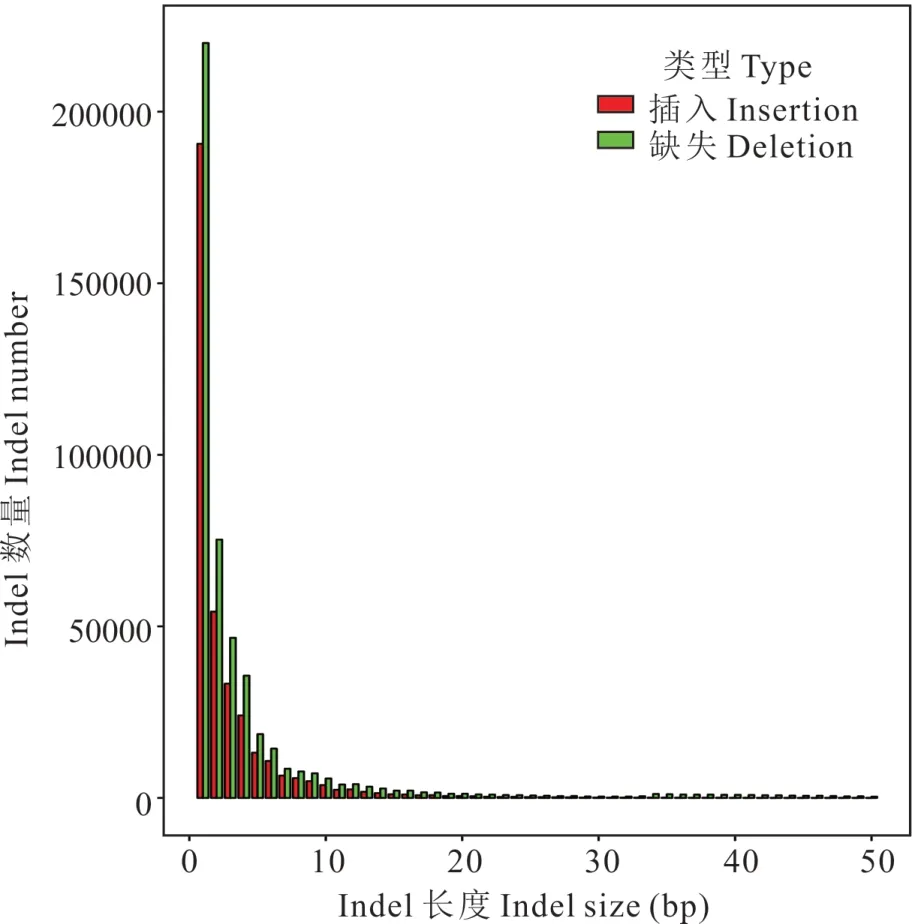
图4 Indels 长度统计Fig.4 Indels length statistics
2.2 群体遗传多样性
2.2.1 牦牛群体的杂合度 9 个牦牛群体的观测杂合度差异较小,在0.2172~0.2440 之间,均低于期望杂合度(表3)。九龙牦牛杂合度最低为0.2172,遗传多样性最贫乏。亚丁牦牛的观测杂合度较低为0.2186,遗传多样性贫乏;拉日马牦牛观测杂合度为0.2233,低于9 个群体平均观测杂合度0.2276,遗传多样性偏低。
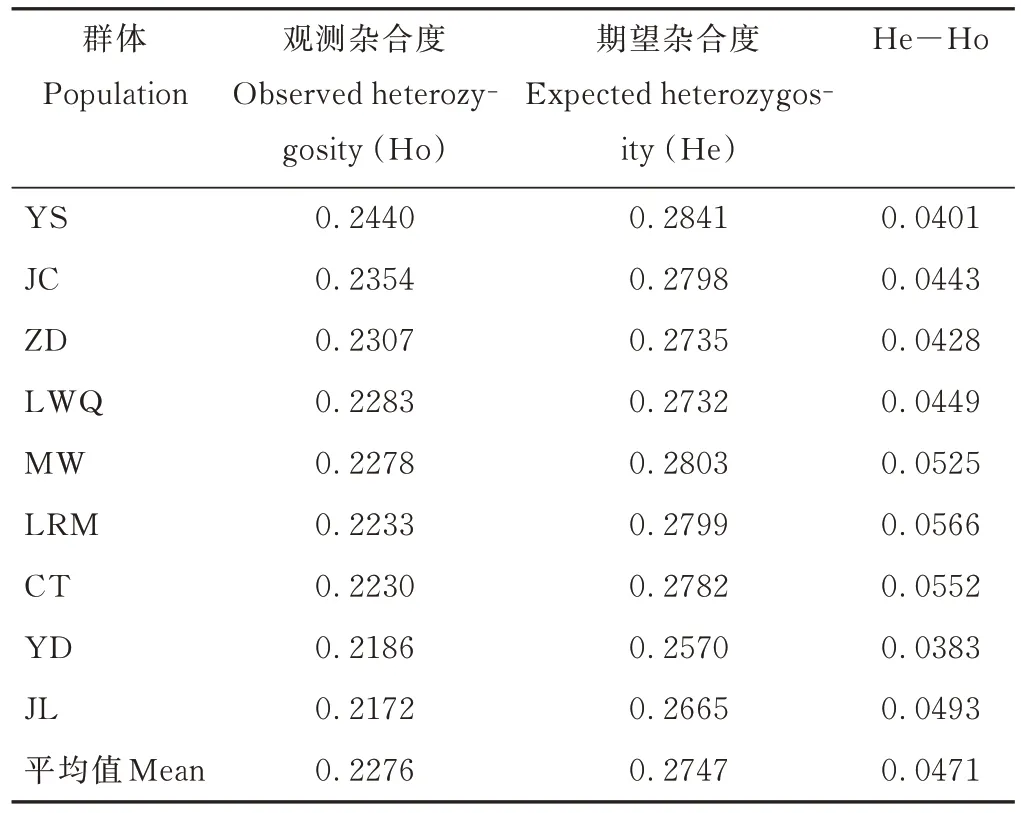
表3 9 个牦牛群体的杂合度Table 3 Heterozygosity of 9 yak populations
2.2.2 牦牛群体的连锁不平衡 9 个群体SNP 距离在0~50 kb 范围内的LD 衰减速度较快,LD 系数约从 0.5 降至 0.2 以下,SNP 距离在 50~300 kb 范围内LD 水平较低在 0.1~0.2 之间,显示在 0~50 kb 范围内各等位基因连锁不平衡程度较高。当LD 系数r2=0.2 时,各群体对应LD 衰减距离基本相等,说明各群体LD 衰减速度差异不大(图5)。
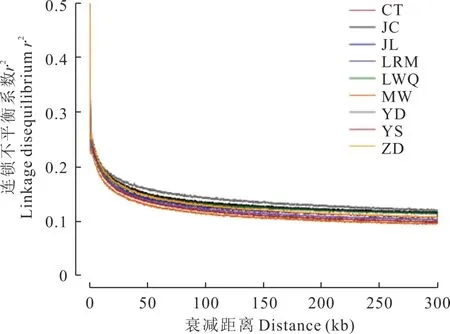
图5 LD 随距离增长的衰减大小Fig.5 Attenuation of LD with distance
2.3 牦牛群体间的遗传关系
通过Pairwise Fst 统计分析不同地方牦牛群体间的遗传距离。亚丁牦牛、九龙牦牛、中甸牦牛以及类乌齐牦牛平均分化指数高于0.05(表4),遗传分化程度较大;拉日马牦牛、麦洼牦牛、金川牦牛、昌台牦牛以及玉树牦牛平均分化指数低于0.05,遗传分化程度较低。亚丁牦牛平均分化指数最高为0.0653,与其他8 个群体存在中等程度(0.05<Fst<0.15)的遗传分化;拉日马牦牛的平均分化指数最低为0.0443,与玉树牦牛、麦洼牦牛群体间的遗传分化程度最小。
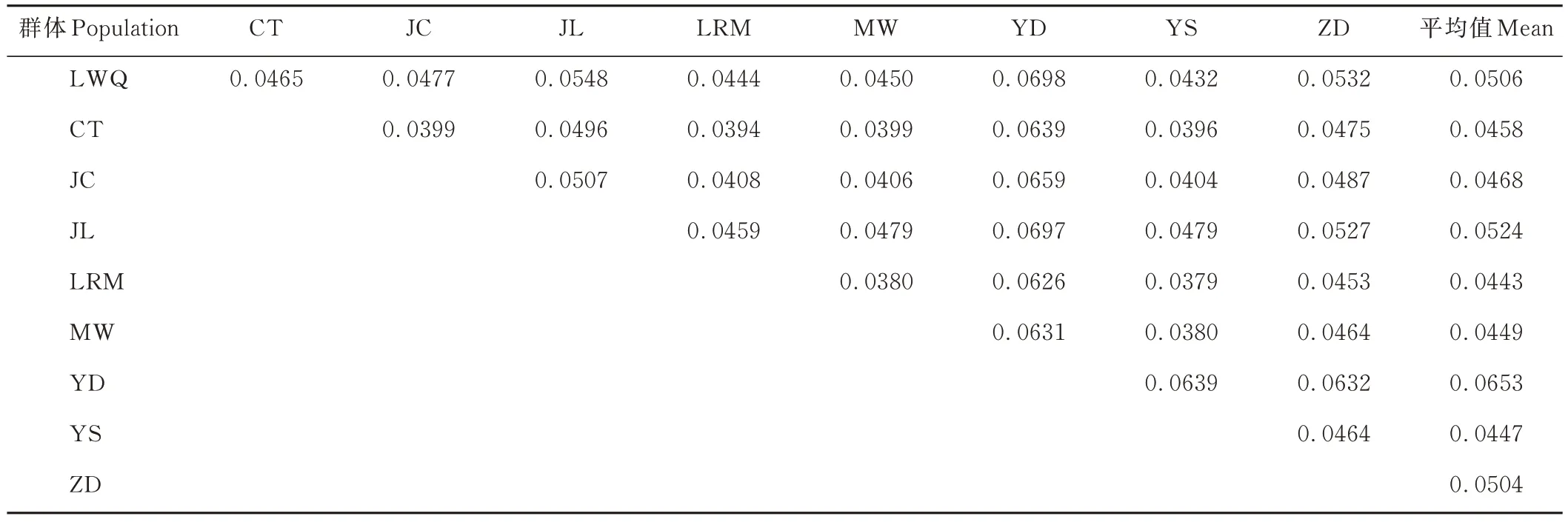
表4 群体间Pairwise FstTable 4 Pairwise Fst between population
基于个体基因组SNP 差异程度,按主成分将不同个体聚类成不同亚群(图6)。亚丁牦牛群体明显与其他地方牦牛群体分开,而且分布距离较远,与Fst 统计结果一致,遗传分化程度较高。在PC2 负值方向,有2个拉日马牦牛样本(LRM-4、LRM-7)与九龙牦牛群体聚集,在 PC2 正值方向,金川牦牛 JC-3、JC-4 样本、麦洼牦牛(MW-4、MW-5、MW-6、MW-8)样本分别独立成群,遗传变异现象明显,其余个体以及拉日马牦牛、昌台牦牛、玉树牦牛、类乌齐牦牛群体间出现严重重叠,亲缘关系较近。
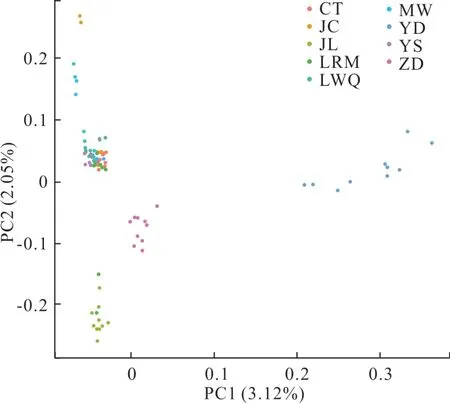
图6 9 个牦牛群体的主成分分析Fig.6 Principal components analysis of 9 yak populations
为了解9 个地方牦牛群体间的亲缘关系,通过最大似然法构建系统进化树(图7),结果显示亚丁牦牛与中甸牦牛亲缘关系最近,拉日马牦牛虽有3 个样本(LRM-4、LRM-7、LRM-6)与九龙牦牛聚为一支,其余7 个样本聚集明显,先与麦洼牦牛聚为一支,说明拉日马牦牛与九龙牦牛存在基因交流,与麦洼牦牛亲缘关系近。类乌齐牦牛与玉树牦牛分群明显,亲缘关系近。金川牦牛和昌台牦牛聚为一支,无明显分群,存在严重基因交流现象。
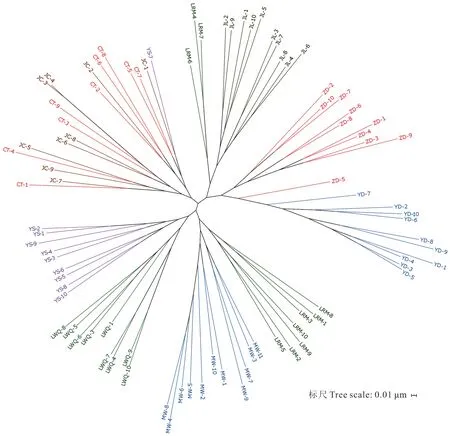
图7 最大似然法构建系统进化树Fig.7 Phylogenetic tree construction by maximum likelihood method
2.4 牦牛群体的遗传结构
对牦牛群体进行群体结构分析,确定最佳分群情况,揭示各牦牛群体的血统构成,以及每个个体的血统潜在来自哪些群体以及对应比例(图8),可追溯其品种的形成历史及亲缘关系。结果显示从K=2~9,亚丁牦牛始终分为一个独立群体,分化程度最高,与Fst 统计、PCA 分析结果一致。Inlikelihood 计算结果显示(图 9),当K=3 时,ΔK值最高,即 9 个地方牦牛群体88 个个体可分为3 个群体:亚丁牦牛、九龙牦牛、其他7 个地方群体(拉日马牦牛、昌台牦牛、金川牦牛、类乌齐牦牛、麦洼牦牛、玉树牦牛、中甸牦牛)为一个群体,最接近牦牛群体的理论分群情况。亚丁牦牛、九龙牦牛血统纯正,仅部分个体出现极少量混杂现象,其他7 个群体血统组成相似。中甸牦牛每个个体均含少量(≤25%)亚丁牦牛和九龙牦牛血统,存在基因交流现象,与亚丁牦牛、九龙牦牛亲缘关系近,与系统进化树分析结果一致。K=3~9,拉日马牦牛血统中含有一定比例九龙牦牛血统,K=6 时起,麦洼牦牛部分个体(MW-4、MW-5、MW-6、MW-8)被归为一个独立群体,与PCA 分析结果一致,拉日马牦牛含有一定比例麦洼牦牛血统,说明拉日马牦牛与九龙牦牛、麦洼牦牛均存在基因交流,与麦洼牦牛血统组成较相似,亲缘关系较近,与系统进化树分析结果一致。

图8 牦牛群体结构堆叠分布Fig.8 Groups structure clustering distribution in yak
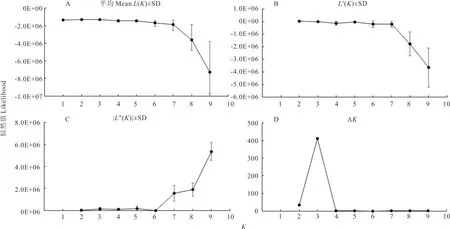
图9 运用Inlikeliood 预估最佳K 值的4 个步骤Fig.9 Four steps to estimate the best K value with inlikelihood
3 讨论
关于牦牛群体遗传多样性已开展大量的研究,柴志欣等[25]利用RAPD 分子标记分析西藏地区11 个牦牛类群的遗传多样性并进行分类;Zhang 等[26]利用16 种微卫星标记分析中国9 个牦牛品种的种群内遗传变异水平;郭松长等[27]利用线粒体DNAD-loop 区部分序列对我国10 个家牦牛品种(类群)进行遗传多样性及分类研究。亚丁牦牛和拉日马牦牛是肉乳兼用型优良地方牦牛资源,探究其分子遗传背景,为开展亚丁牦牛和拉日马牦牛遗传改良和育种提供理论依据。
本研究利用RAD 测序技术在全基因组水平探究亚丁牦牛和拉日马牦牛的遗传多样性及遗传结构。群内遗传多样性分析结果显示,9 个牦牛群体的观测杂合度为0.2172~0.2440,均低于期望杂合度,说明各地方群体内存在一定程度近交率[28]。其中,麦洼牦牛、九龙牦牛、昌台牦牛、类乌齐牦牛和中甸牦牛的观测杂合度均低于运用微卫星标记所测群体观测杂合度(0.5650~0.9599)[29−32],微卫星标记所测的杂合度较高与其标记位点多态性高、分布集中有关,RAD 简化基因组测序较全面地反映整个基因组多态位点情况[9]。亚丁牦牛和拉日马牦牛的观测杂合度分别为0.2186 和0.2233。亚丁牦牛遗传多样性贫乏,可能因为亚丁地区背山封闭,近亲交配严重,导致繁殖性能降低,从而引起群体内个体杂合性降低[5]。拉日马牦牛遗传多样性偏低,推测其群内存在一定程度近交率。9 个群体间LD 衰减速度差异较小,可能与各群体间遗传多样性差异小有关[33]。
本研究中亚丁牦牛的平均遗传分化指数Fst 最高为0.0653,与其他8 个地方牦牛群体存在中度分化,群体结构分析显示从K=2~9 亚丁牦牛始终分为一个独立群体,血统构成纯正,且Inlikelihood 计算结果显示亚丁牦牛理论上应归为一个独立群体。生物的进化以自然环境为选择主线,不断修正和改变基因的频率,从而引导生物的进化方向[34]。据史料记载,亚丁地区饲养牦牛历史悠久,公元619−786 年,藏王将吐蕃国牦牛迁徙至东义片区即现在的亚丁地区饲养,该区域受赤土河、东义河切割,山高谷深,立体气候明显,亚丁牦牛在对环境的适应过程中同时受到人工选择的作用,亚丁地区和中甸地区历史上曾有相互交换种牦牛和在交界地混牧的习惯[6],因此中甸牦牛血统中含有少量亚丁牦牛血统,具有亲缘关系,经过长期的自群繁育,亚丁牦牛群体血统纯正,与其他地方群体间产生了遗传变异。总之,亚丁牦牛虽体遗传多样性贫乏,但血统纯正,其遗传背景与其他8 个地方群体差异明显,肉奶性能良好,其产奶量居中国牦牛之最,建议亚丁牦牛单独列为一个牦牛品种进行开发利用和保护。
拉日马牦牛的平均遗传分化指数Fst 最低为0.0443,群体遗传背景较复杂,与九龙牦牛和麦洼牦牛均存在基因交流,与麦洼牦牛亲缘关系最近。拉日马牦牛与九龙牦牛中心产区相距较远,存在基因交流的原因可能是九龙牦牛向拉日马镇发生过迁徙,九龙牦牛属世界上最好的种牛,存在人工引种的可能。新龙县志办记载,在1925 年前后,瓦西洛尔龙作为当时新龙县拉日马麦洼部落头人(称“麦本”),因部落内部纠纷,带着牲畜7000 多头(主要是牦牛)背井离乡前往现阿坝州藏族羌族自治州红原县境内麦洼区定居。据文献资料记载及牦牛科研工作者多次调查,麦洼牦牛来自甘孜藏族自治州北部色达、德格、炉霍、新龙等县[6]。本研究在全基因组水平上进一步证明拉日马牦牛与麦洼牦牛存在基因交流,属麦洼牦牛的祖先群之一,拉日马牦牛较麦洼牦牛在肉奶性能方面存在一定的优越性,其净肉率和产奶量均高于麦洼牦牛[8−9]。
4 结论
本研究以亚丁牦牛和拉日马牦牛及其他7 个地方牦牛类群为研究对象进行SNP 检测和群体遗传关系分析。亚丁牦牛遗传多样性贫乏,血统纯正,与其他8 个地方牦牛群体存在中度分化,可列为一个独立遗传资源进一步研究和保护。拉日马牦牛遗传多样性偏低,属麦洼牦牛的祖先群之一,其群体遗传结构较复杂,需结合其他研究方法、增加样本量进一步开展研究工作。
