微RNA风险评分模型预测肝细胞癌预后的价值分析
2021-05-17黄秀红谢肖立姜慧卿
黄秀红,谢肖立,姜慧卿
河北医科大学第二医院 消化内科,石家庄 050000
肝细胞癌(HCC)是最常见的原发性肝癌,其发病率在所有恶性肿瘤中排第6位,致死率排第4位,世界卫生组织估计,截至2030年,全球每年将有超过100万死于HCC的患者[1-2]。由于HCC起病隐匿,进展迅速且异质性高,尽管外科手术、射频消融、化学栓塞甚至肝移植已广泛用于HCC治疗,但HCC患者的存活率仍然较低[3-4]。由于与HCC发生和发展有关的生物学过程非常复杂,迄今为止尚无敏感高效的预后生物标志物,因此,有必要探索新的能够高效预测HCC预后的生物标志物或模型,用于HCC的诊断、预后和治疗。
微RNA(microRNA,miRNA)是一类参与mRNA转录后调控的非编码小RNA,通过与目标mRNA的3′非编码区结合,抑制转译或降解mRNA,继而在转录后水平调控靶基因的表达,从而影响细胞生物学功能,miRNA的差异表达在多种恶性肿瘤的发生发展中发挥关键作用[5-6],而且多项研究[7-9]已经确定miRNA模型能够预测恶性肿瘤患者的预后,比如乳腺癌、前列腺癌及卵巢癌。而在本次研究中,首先从TCGA中下载HCC基因及临床数据,随后找出与HCC预后相关的miRNA,构建预测HCC预后的miRNA风险评分模型,并验证其检验效能。
1 资料与方法
1.1 数据下载与整理 从TCGA数据库(https://portal.gdc.cancer.gov/)中下载HCC miRNA转录组数据及临床数据。
1.2 差异分析 利用R语言的limma包对HCC与正常肝组织之间的miRNA进行基因差异表达对比分析,并设置差异倍数log2FC(fold change,FC)>1.0且P<0. 01。
1.3 miRNA风险评分模型的构建 从临床数据中提取生存信息,包括生存时间和生存状态,与差异表达的miRNA进行整合,利用R语言caret包将其随机分成训练集和测试集,利用survival、glmnet、survminer包对训练集进行单因素Cox回归分析,并设定P<0.01为差异具有统计学意义。然后通过10倍交叉验证的LASSO-Cox回归分析进一步筛选预后miRNA,并计算每个miRNA的危险比(HR)和回归系数,基于miRNA表达水平及回归系数的线性组合构建与HCC预后相关的风险评分模型。
1.4 miRNA风险评分模型的验证 根据风险评分模型计算出训练集中每个样本的风险得分,并根据中位风险得分值将样本分为高风险组和低风险组,采用log-rank检验的Kaplan-Meier生存曲线评估高风险组与低风险组的预后差异,组间差异比较时设定P<0.05为差异具有统计学意义,同时在测试集中评估其稳健性。为了评估该模型是否可以预测同一临床分期的患者预后,对不同TNM分期的患者进行了分层分析,将HCC患者分为低TNM组(Ⅰ和Ⅱ期)和高TNM组(Ⅲ和Ⅳ期),同样采用log-rank检验的Kaplan-Meier生存曲线评估高TNM组和低TNM组的预后差异,并设定P<0.05为差异具有统计学意义。
1.5 miRNA风险评分模型的评估 通过R语言的survival ROC包绘制ROC曲线,计算出ROC曲线下面积(AUC),并与传统TNM分期比较,评估该模型的预测准确性,其中AUC> 0.7被认为具有良好的预测效能。最后对风险评分模型和临床特征进行单因素和多因素Cox回归分析评估该模型预后独立性,并设定P<0.05为差异具有统计学意义。
2 结果
2.1 一般资料 miRNA样本共425例,包括375例HCC样本及50例癌旁样本,并将临床数据中生存时间、生存状态及TNM分期缺失的数据删除,获得临床数据352例(表1)。

表1 TCGA数据库HCC患者的临床数据
2.2 差异表达的miRNA 利用R语言对HCC及癌旁组织进行miRNA差异表达分析,以log2FC>1.0 且P<0.01 为标准,共筛选出300个差异表达miRNAs,上调基因260个,下调基因40个。
2.3 miRNA风险评分模型的构建 将差异表达的miRNA与临床数据进行整合匹配,共得到347个样本,利用R语言将其按照1∶1随机分成训练集(n=175)和测试集(n=172),对训练集依次进行单因素Cox及10倍交叉验证的LASSO-Cox回归分析。单因素Cox回归分析显示,23个miRNAs与HCC预后相关(P<0.01)。然后通过LASSO回归进一步筛选miRNA,结果显示9个miRNAs: hsa-miR-139-5p、hsa-miR-1180-3p、hsa-miR-4652-5p、hsa-miR-1269b、hsa-miR-122b-5p、hsa-miR-3677-5p、hsa-miR-3680-3p、hsa-miR-509-3-5p、hsa-miR-31-5p与HCC预后显著相关(图1)。
最后将LASSO回归筛选出的miRNAs拟合到多因素Cox回归分析,最终确定了包括hsa-miR-139-5p、hsa-miR-1180-3p、hsa-miR-1269b、hsa-miR-3680-3p、hsa-miR-509-3-5p、hsa-miR-31-5p在内的6个miRNAs,构建风险评分公式并绘制森林图(图2)。风险评分公式:hsa-miR-139-5p×(-0.001 07)+hsa-miR-1180-3p×0.001 57+hsa-miR-1269b×0.000 26+hsa-miR-3680-3p×0.162 79+hsa-miR-509-3-5p×0.002 46+hsa-miR-31-5p×0.010 67,计算每个样品的风险得分。在这些miRNAs中,hsa-miR-1180-3p、hsa-miR-1269b、hsa-miR-3680-3p、hsa-miR-509-3-5p、hsa-miR-31-5p的系数为正,表明这些基因表达水平越高,生存期越短,而hsa-miR-139-5p的系数为负,表明该基因表达水平越高,生存期越长。
2.4 miRNA风险评分模型的验证 根据miRNA风险评分模型,计算训练集中每个样本的风险得分,再根据中位风险得分值(n=0.918 637 4)将训练集分为高风险组(n=87)和低风险组(n=88)。生存曲线显示(图3a)高风险组患者的生存率明显低于低风险组,差异具有统计学意义(P<0.05)。同样地,根据相同的风险评分模型及中位风险得分值(n=0.918 637 4),将测试集中的患者分为高风险组(n=81)和低风险组(n=91),生存曲线显示(图3b),高风险组患者的生存率明显低于低风险组,差异具有统计学意义(P<0.05)。
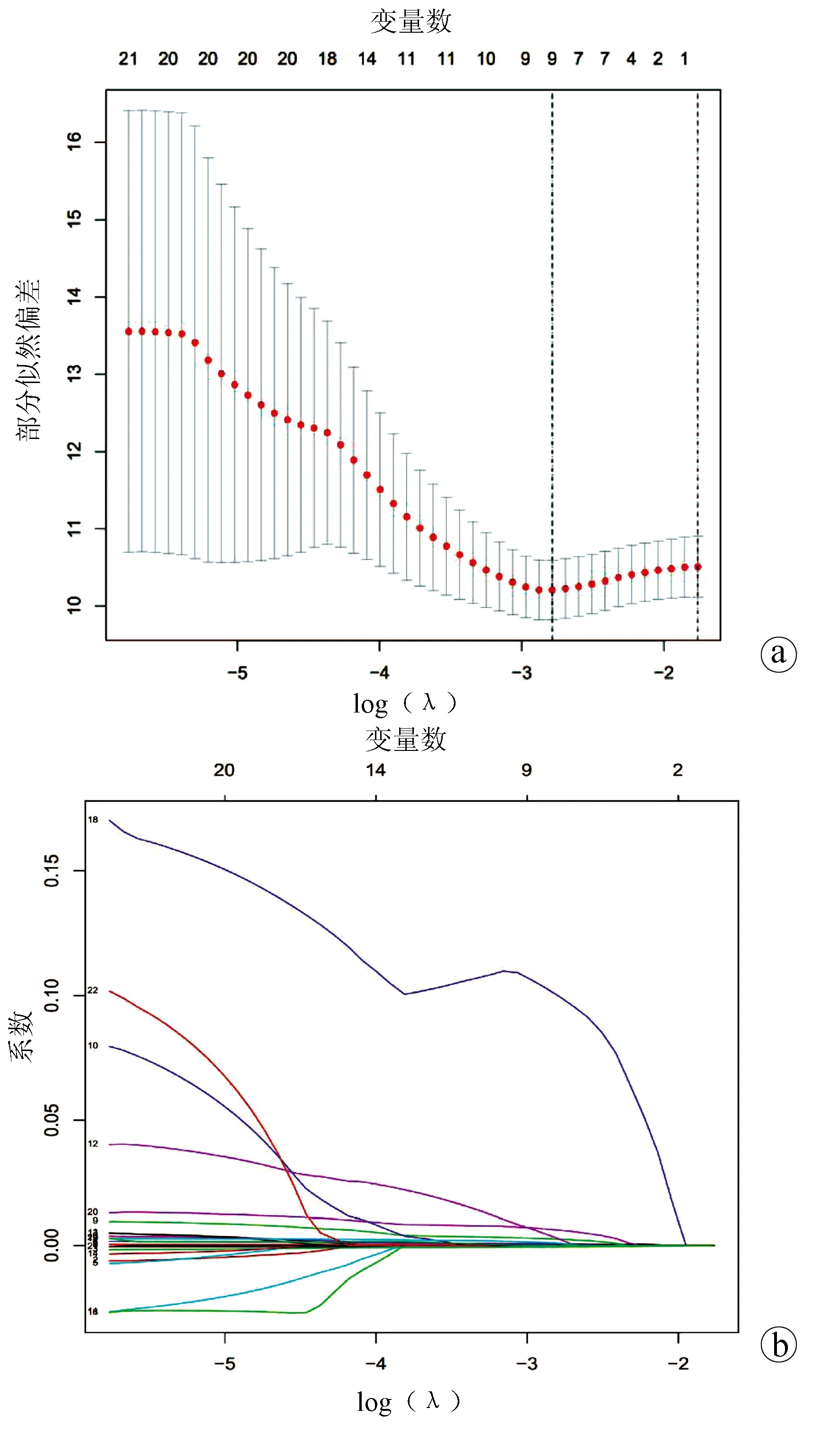
注:a,LASSO筛选与生存相关的miRNAs;b,与HCC患者预后相关的miRNAs的LASSO系数。

注:**,P<0.01;***,P<0.001。
随后根据样本的风险得分排名,分别绘制训练集和测试集的风险图、生存状态图及与HCC预后相关的6个miRNAs的风险热图(图4、5)。结果表明高风险评分的患者预后较差,且系数为正的miRNAs(hsa-miR-1180-3p、hsa-miR-1269b、hsa-miR-3680-3p、hsa-miR-509-3-5p、hsa-miR-31-5p)表达上调,而低风险评分的患者预后较好,且系数为负的miRNA(hsa-miR-139-5p)表达上调。

注:a,训练集生存曲线;b,测试集生存曲线。
将患者按TNM分期分为低TNM组(Ⅰ和Ⅱ期)和高TNM组(Ⅲ和Ⅳ期),Kaplan-Meier曲线显示低TNM组、高TNM组的高风险患者的生存率显著低于低风险患者(P<0.05),表明该模型可以预测同一临床分期的患者预后。

注:a,训练集风险图;b,训练集生存状态图;c,训练集风险热图

注:a,测试集风险图;b,测试集生存状态图;c,测试集风险热图。
2.5 miRNA风险评分模型的评估 利用R语言绘制miRNA模型及TNM分期的ROC曲线并计算AUC,结果显示训练集(图7a)中,miRNA模型及TNM分期的AUC分别为0.817、0.667,测试集(图7b)分别为0.808、0.665,及合集样本(图7c)分别为0.814、0.663,miRNA模型预测准确性均优于TNM分期。
将年龄、性别、肿瘤分级、TNM分期、miRNA评分分别进行单因素、多因素Cox独立预后分析,单因素分析结果显示T分期、M分期、TNM分期及miRNA评分是HCC患者预后的相关因素(P值均<0.05)(图8a),多因素分析结果显示miRNA评分模型可作为HCC的独立预后因子(P<0.05)(图8b)。

注:a,低TNM组(Ⅰ和Ⅱ期)生存曲线;b,高TNM组(Ⅲ和Ⅳ期)生存曲线。

注:a,miRNA模型和TNM分期在训练集的ROC曲线;b,miRNA模型和TNM分期在测试集的ROC曲线;c,miRNA模型和TNM

注:a,单因素分析;b,多因素分析。
3 讨论
原发性肝癌是常见的恶性肿瘤,在各个年龄段均可发病,发病率和死亡率比较高,预后很差。目前,用于预测HCC预后主要依据传统的TNM分期[10],尽管TNM分期在恶性肿瘤的诊断和治疗中起着重要作用,但由于肿瘤的异质性和个体差异,其无法反映内部的生物学过程和疾病进展,因此有必要寻找一种新的能够高效预测HCC预后的生物标志物或模型,辅助临床工作。
在本次研究中,首先从TCGA中下载HCC的miRNA表达量及临床数据,TCGA是由美国国立卫生研究院支持发起的,包括30多种癌症,旨在提供不同癌症的全面基因分析并建立与临床结果的相关性,这些数据对于癌症的研究具有巨大的潜力,并且已经利用这些数据取得了许多成果[11-16]。当作者试图从训练集中确定预后特征时,由于样本量小,基因数量众多,因此对训练集中的数据进行了10倍交叉验证的LASSO-Cox回归分析。其中LASSO是一种创新的回归变量选择方法,在系数的绝对值之和小于一个常数的情况下将残差平方和最小化,从而使某些回归系数严格等于0,从而选择出对因变量影响较大的自变量,是通过构造一个惩罚函数得到一个较为精炼的模型[17],从而减少模型的过度拟合。而交叉验证法是比较常用的推测估计惩罚系数λ的方法,而λ值最终是由使平均交叉验证误差最小的最小化λ确定的。然后根据Cox多因素回归分析,得出和HCC预后相关的6种miRNAs。通过回顾已发表的文献[18-20],作者发现 miRNA的失控与HCC的发生、耐药、预后相关,广泛参与HCC中肿瘤抑制基因的失活和癌基因的激活。其中hsa-miR-1180-3p、hsa-miR-1269b、hsa-miR-3680-3p、hsa-miR-509-3-5p、hsa-miR-31-5p的表达在HCC组织中上调,可能起致癌作用,hsa-miR-139-5p在HCC组织中的表达下调,可能起抑癌作用,而hsa-miR-139-5p可以通过减少SLITRK4的表达抑制HCC细胞的生长[21]。然后基于回归系数构建了风险评分模型,计算出训练集中每位患者的风险得分,然后根据中位风险得分值将其分为高风险组和低风险组,并在测试集中验证了该miRNA风险评分模型的预后价值。生存曲线分析表明,训练集、测试集的高风险组和低风险组的生存曲线均观察到明显的分离(P值均<0.05)。为了检验该模型预测效能,绘制了ROC曲线,并计算了相应的AUC,将其与传统TNM分期比较,结果显示在训练集、测试集及合集样本中miRNA模型的预测准确性均优于TNM分期。最后将年龄、性别、分级、分期、miRNA评分模型分别进行单因素、多因素Cox独立预后分析,结果显示该miRNA评分模型可作为HCC的独立预后因子。该模型与传统TNM分期相比较,能够更好地反应内在生物学进程和疾病进展,受主观因素影响少,且该预测模型具有可评估性和可重复性等优点,可与传统TNM分期互补,共同应用于临床工作。同时本研究也有一定的不足,所有分析均通过TCGA数据库完成,以后可同时设置内部验证集和外部验证集(如GEO数据库)验证该模型预测准确性,而且未进行相关实验,这些都有待进一步研究工作。
利益冲突声明:本研究不存在研究者、伦理委员会成员、受试者监护人以及与公开研究成果有关的利益冲突。
作者贡献声明:黄秀红负责酝酿和设计实验,下载、分析数据,统计分析,论文撰写;谢肖立负责协助论文修改;姜慧卿负责研究指导、论文修改。
