运动介导线粒体未折叠蛋白反应调控线粒体质量控制机制研究进展
2021-05-15马春伟高炳宏
马春伟 高炳宏
1 上海体育学院运动科学学院(上海200438)
2 运城学院体育系(山西运城044000)
3 上海体育学院体育教育训练学院(上海200438)
线粒体是高度动态和半自主的细胞器,在生理代谢、应激反应和细胞死亡等多种细胞功能的管理中发挥重要作用,维持线粒体稳态对于细胞生存、协调应激反应和介导细胞死亡至关重要。为了维持线粒体稳态,细胞产生新的线粒体和去除受损或冗余的线粒体均需要精确的控制过程[1],线粒体稳态受到破坏,如线粒体聚集、信号转导通路异常或应激反应导致的线粒体功能障碍,是包括神经退行性病变、心血管疾病和癌症在内的诸多疾病发生的根源[2]。线粒体质量控制是维持线粒体稳态的有效手段,包括线粒体内蛋白质稳态、线粒体生物发生、线粒体融合与分裂、线粒体自噬等生理过程,参与调节线粒体的形态、体积和功能,是细胞应激反应的关键组成部分[3,4]。
线粒体内蛋白质的稳定是维持线粒体形态、功能的前提和基础,为了将线粒体功能整合到细胞网络中,需要通过一系列的信号转导通路来监控线粒体适应程度,使线粒体功能与细胞需求协调一致。在应激条件下,信号转导通路受损,线粒体内外会聚集大量的未折叠蛋白或错误折叠蛋白,导致蛋白毒性应激,最终导致细胞功能下降甚至死亡[5]。应对诸如缺氧、氧化应激等外界环境变化时,细胞利用受损的蛋白质输入作为线粒体功能障碍的传感器,激活线粒体未折叠蛋白反应(mitochondrial unfolded protein response,UPRmt)[6],这是细胞应激后产生的一种适应性反应,它维持着线粒体的蛋白平衡,介导组织间的信号传导,调控着机体的衰老[7]。运动是一种典型的应激,不同的运动方式会引起线粒体内不同程度的未折叠蛋白反应,进而调控线粒体质量控制机制。虽目前已经对UPRmt展开了一系列的研究,但研究的关注点仍停留在细胞层面或线虫等低等动物上,对哺乳动物体内的UPRmt机制的研究仅处于起步阶段,阐明哺乳动物中UPRmt与线粒体质量控制之间的关系,以及运动在这一关系网络中扮演了何种角色等问题,对于理解线粒体在不同应激状态下发生的适应性反应尤为重要,并最终为靶向治疗及延长生命跨度提供新的途径[8]。
1 线粒体未折叠蛋白反应与线粒体质量控制概述
1.1 线粒体未折叠蛋白反应
线粒体功能的多样性和适应性取决于蛋白质、蛋白质复合物和应激反应途径的协同作用[6],线粒体基因组包含1000 多种蛋白质[9],其中有13 种蛋白质在线粒体内编码,是呼吸链或ATP 合成酶的组成成分,剩余99%的线粒体蛋白在细胞核内编码,在细胞质内作为前体合成,并通过专门的蛋白转位酶导入线粒体[10],这种复杂的蛋白质输入机制可有效维持线粒体的基本功能[11]。为有效维持蛋白质稳态,细胞核与线粒体之间拥有双向的信号通路[12]:一方面,通过信号转导机制由细胞核协调完成,以满足细胞的能量需求,这种通信称为顺行信号调节;另一方面,线粒体还可向细胞核转导信号,调节核编码基因的遗传表达,这种现象称为逆行反应信号,这种信号主要作用是协调细胞核基因表达的适应性变化,以减少或解决线粒体应激等问题[13],UPRmt就是典型的逆行反应信号转导过程。
UPRmt在遗传或药理学上触发线粒体应激时,可以感知线粒体蛋白质组的功能障碍并将其传递至细胞核,激活修复损伤的转录过程。另外,UPRmt也是一种自适应信号通路,借助线粒体到细胞核的信号转导通路,检测线粒体内的蛋白毒性应激,诱导线粒体分子伴侣和蛋白酶等线粒体保护因子,并通过调控基因表达过程恢复细胞器内的蛋白平衡,维持或重建线粒体蛋白折叠环境中的蛋白质稳态,恢复线粒体功能[14]。UPRmt可同步线粒体和核基因组的活性,通过激活线粒体蛋白质量控制网络调控线粒体蛋白质稳态,从而确保线粒体蛋白质组的质量,还通过将代谢类型从依赖线粒体氧化磷酸化转移到细胞质糖酵解,减轻线粒体的应激需求[15]。在应激状态下,线粒体未折叠蛋白积累可导致线粒体结构和功能发生改变,线粒体短期轻度的应激可激活UPRmt反应,启动细胞内防御性应答系统,抵抗线粒体损伤,维持或促进线粒体功能的发挥;而线粒体应激水平过高,UPRmt的保护作用不足以抵抗线粒体的损伤程度时,则会通过介导细胞凋亡,加重细胞的不可逆损伤[16]。UPRmt主要通过诱导控制蛋白质折叠、组装和降解的线粒体伴侣蛋白和蛋白酶的表达,来维持线粒体蛋白稳定[13],伴侣蛋白包括热休克蛋白60(heat shock protein 60,Hsp60)和线粒体热休克蛋白70(mitochondrial heat shock protein 70,mtHsp70)等可促进蛋白质折叠并防止聚集体形成,蛋白酶主要包括m-AAA 和i-AAA 蛋白酶,可以消除受损或错误折叠的蛋白质的积累[6]。目前已知的哺乳动物UPRmt信号转导途径主要包括活化转录因子4(activating transcription factor 4,ATF4)/活化转录因子5(activating transcription factor 5,ATF5)-C/ EBP同源蛋白(C/EBP homologous protein,CHOP)途径、沉默信息调节因子3(silent information regulator factor 2 related enzyme 3,Sirt3)-叉头框蛋白3a(forkhead box protein O3a,FOXO3a)-锰超氧化物歧化酶(manganese superoxide dismutase,superoxide dismutase 2,SOD2)途径、蛋白激酶B(protein Kinase B,AKT)/雌激素受体(estrogen receptor,ERα)/高温需求蛋白A2(high temperature requirement protein A2,HtrA2,也称为OMI)途径[17]。
1.2 线粒体质量控制
线粒体质量控制包括一系列相互关联且相互牵制的生理过程。新线粒体的合成是一个复杂的过程,称为线粒体生物发生,涉及线粒体外膜和内膜的膜合成、线粒体DNA 复制以及在细胞质中合成的蛋白质输入并运输到适当的线粒体内区室(外膜、膜间隙、内膜或基质)等过程[6],线粒体的生物发生可以促进现有线粒体网络的扩展。其中,过氧化物酶体增殖因子激活因子受体-共激活因子1α(peroxisome proliferator-activated receptor gamma coactivator 1 alpha,PGC-1α)和线粒体转录因子A(transcription factorA,Tfam)等因子参与诱导线粒体的生物发生。线粒体通过融合和分裂这两个相反的过程不断的进行重构,构成线粒体动力学。一方面,线粒体融合可以使完整的线粒体与轻微功能障碍的线粒体混合,保护细胞器不被降解,并进行物质交换,同时取代受损的线粒体DNA,恢复线粒体完整性。在哺乳动物细胞中,除了视神经萎缩蛋白1(opticAtrophy protein 1,Opa1)外,线粒体融合蛋白1/2(mitochondrial fusion protein 1/2,Mfn1/2 )也是调节融合过程的线粒体膜蛋白,它们分别是促进线粒体内外膜协调融合的重要因素,融合过程促进线粒体蛋白、脂质和线粒体DNA 的迁移。另一方面,通过线粒体分裂分离出不可逆的受损线粒体,并被线粒体自噬消除。线粒体膜分裂是主要由动力相关蛋白1(dynamin-related protein-1,Drp1)与分裂蛋白1(fission protein 1,Fis1)联合介导。线粒体分裂可以定位到特定的网状损伤区域,确保选择性的去除网状损伤区域[18,19]。线粒体动力学受同源性磷酸酶张力蛋白诱导的激酶1(PTEN induced putative kinase 1,PINK1)-Parkin(线粒体自噬相关蛋白,是一种E3 连接酶)通路的调控,这一调控通路是线粒体自噬的一部分,线粒体自噬可以特异性消除受损严重的线粒体。Somak Das 通过激光共聚焦显微技术观察到线粒体分裂先于线粒体自噬发生,PINK1/Parkin 可能通过阻断融合而促进分裂,从而促进线粒体自噬[20],除PINK1/Parkin 通路外,线粒体自噬通路还包括BCL2/腺病毒E1B-19kDa 相互作用蛋白3(BCL2/adenovirus E1B 19-kDainteracting protein 3,BNIP3)-BCL2/腺病毒E1B-19kDa 相互作用蛋白3 样(BCL2/adenovirus E1B 19-kDainteracting protein 3-like,Bnip3L,也称为Nix)通路和FUN14域蛋白1(FUN14domain containing1,FUNDC1)通路[21,22]。综上,细胞通过线粒体生物发生、线粒体自噬(靶向降解)、线粒体动力学(融合与分裂)等过程维持线粒体稳态[23],虽然其内在机制仍未被完全阐明,但这一过程是细胞存活所必需的,也是维持健康的线粒体池所必需的,尤其是在运动等应激状态下可以更大限度地满足细胞代谢需要[24]。
2 线粒体质量控制与线粒体未折叠蛋白反应的相关关系
2.1 线粒体未折叠蛋白反应直接调节线粒体RNA 的加工过程
UPRmt涉及核基因的广泛诱导过程,包括参与折叠基质定位蛋白、前体RNA 加工和翻译[25]。线粒体DNA编码的转运RNA(transfer RNA,tRNA)对于线粒体内的翻译和RNA 成熟至关重要。转录后,线粒体tRNA在5’和3’端被核糖核酸酶P(Ribonuclease P,RNase P)和核糖核酸酶Z(Ribonuclease Z,RNase Z)复合物加工[26]。人类的RNase P由线粒体RNase P亚基(mitochondrial RNase P subunit,TRMT10C/MRPP1)、短链氧化还原酶羟类固醇17β-脱氢酶10(short-chain oxidoreductase hydroxysteroid 17β- dehydrogenase 10,HSD17B10/MRPP2)和金属核酸酶(metallonuclease,KIAA0391 / MRPP3)组成。在线粒体应激状态下,UPRmt激活会因LON 蛋白酶降解增加而导致MRPP3 降低[25]。因此,UPRmt激活通过MRPP3 降解使真核起始因子2α(eukaryoticinitiation factor 2α,eIF2α)和线粒体磷酸化,从而减少细胞质内的蛋白质合成,这可能是促进蛋白质稳定的原因之一[6]。也有文献指出:线粒体内发生的翻译抑制过程与转录抑制导致的前体RNA 加工缺陷、LON 依赖的线粒体前体RNA 加工核酸酶MRPP3的更新是同时发生的[25,27]。
2.2 线粒体自噬与线粒体未折叠蛋白反应
在线粒体自噬的PINK1/Parkin 通路中,如果线粒体应激时PINK1 不能通过线粒体内膜输入,它会积聚在外膜上,激酶域面向细胞质[6]。PINK1 磷酸化泛素以激活Parkin E3 泛素连接酶,导致多个线粒体外膜蛋白泛素化[28-30],泛素化的线粒体蛋白的积累募集了衔接蛋白,促进自噬装置的靶向识别[31]。基于UPRmt和线粒体自噬的通路都是在线粒体蛋白质输入效率的水平上受到调控的,所以线粒体蛋白质输入有可能是UPRmt与线粒体自噬转换过程中的关键点:当线粒体功能障碍较轻时,细胞可能偏向于UPRmt的激活,此时PINK1 进入线粒体,被加工后释放入细胞质中发生降解。然而,当细胞器功能障碍加重,PINK1 在受损最严重的细胞器外膜上积累时,两种途径都被激活,只有受损最严重的线粒体被靶向降解[32]。也有研究指出:应激状态下,Sirt3-FOXO3a-PINK1-Parkin 通路可能是线粒体融合-分裂-线粒体自噬的信号传导途径,但其机制仍有待于进行深入研究[20]。
哺乳动物自噬的Nix 驱动机制,即外膜蛋白Nix 与微管相关蛋白3(microtubule-associated protein 1 light chain 3,LC3)结合,从而启动自噬[33]。Nix 在缺氧时也会上调[34],这表明Nix 在应激期间通过自噬作用广泛参与线粒体蛋白平衡的恢复,LC3 也成为判断自噬发生的关键指标之一[35]。而UPRmt中的Sirt3/FOXO3通路也通过LC3 参与自噬过程,FOXO3 产生抗氧化反应这一过程的效应包括LC3B 的脂化、多个自噬基因的诱导及自噬率的增加,这也表明自噬和自噬通量受到了刺激[36]。同时,参与UPRmt的蛋白酶酪蛋白裂解酶P(Caseinolytic protease,ClpP)与UPRmt的关系表现在AMPK 磷酸化的增加可促进LC3 转化为LC3-Ⅱ,LC3-Ⅱ也是与UPRmt相关的线粒体自噬标志物[37]。Papa 也证实UPRmt中的Sirt3-FOXO3a 轴在线粒体自噬中发挥作 用[36],Tseng 的研究也得出了一致结论,UPRmt的Sirt3-FOXO3a 信号转导过程激活Bnip3、Nix 和LC3-II/LC3-I,在氧化应激条件下调控线粒体自噬[38]。
在线粒体自噬FUNDC1 通路中,FUNDC1/HSC70(结构性热休克蛋白70)途径促进未折叠的胞质蛋白质的降解,即线粒体外膜蛋白与分子伴侣HSC70 相互作用,以促进胞质中未折叠蛋白的线粒体易位,通过线粒体Lon 蛋白水解酶(Lon peptidase 1,LONP1)降解的非聚集性线粒体相关蛋白聚集体,并以Fis1 依赖性方式与线粒体分离,随后可通过自噬降解。在生理状态下,结构型热休克蛋白70(heat shock cognate70,HSC70)维持蛋白稳定过程中有识别未折叠蛋白的能力,所以可以推测FUNDC1/HSC70 轴可能是活跃的,而转运蛋白将被LONP1 蛋白酶降解。同时,由于FUNDC1 在Ser13位点的去磷酸化作用,调控细胞质中未折叠蛋白的线粒体转位[39]。
综上所述,线粒体自噬的PINK/Parkin 通路、BNIP3/NIX 通路和FUNDC1通路都直接或间接与UPRmt有关,激活UPRmt的许多应激也激活线粒体自噬,因此,有理由相信UPRmt和线粒体自噬是互补的:UPRmt可以作为抵抗线粒体蛋白质损害的第一道防线,而线粒体自噬则用于去除冗余的线粒体[36,40]。
2.3 线粒体生物发生与线粒体未折叠蛋白反应
有研究表明,线粒体膜间腔中存在一个单独的UPRmt信号通路,即Sirt3-FOXO3 通路,该通路专门对未折叠蛋白应激作出应激反应。定位于线粒体膜间腔的内切核酸酶G(Endonuclease G,EndoG)表达改变导致AKT 磷酸化和核激素受体ERα的激活,导致线粒体膜间腔定位的质量控制蛋白酶HtrA2 和转录因子核呼吸因子1(nuclear respiratory factor 1,NRF1)的表达增加,这涉及线粒体生物发生[17]。早前的研究证实UPRmt的Sirt3-FOXO3 轴可上调PGC-1α、Tfam 等因子促进线粒体生物发生。同时,Sirt3 介导的FOXO3 去乙酰化也可促进线粒体生物发生,导致线粒体质量增加[38]。
线粒体未折叠蛋白反应是从线粒体到细胞核的反馈性调节反应,这种逆行信号是有据可循的。线粒体开放阅读框12S rRNA-C(mitochondrial open reading frame of the 12S rRNA-c,MOTS-c)是第一个被识别的线粒体DNA 编码的核转录调控因子,是线粒体和细胞核之间定向通信的最主要手段之一[41],AMP 活化蛋白激酶(AMP-activated protein kinase,AMPK)是已知的唯一与MOTS-c 在功能上发挥相互作用的激酶,AMPK 是线粒体生物发生与自噬的上游共同控制点,抑制或激活AMPK,线粒体的生物发生和自噬均同步发生[42]。同时,AMPK被认为是UPRmt的感受器,应激下AMPK 的抑制对热休克蛋白70(heat shock protein 70,Hsp70)的表达有促进作用,而ClpP 基因沉默可导致AMPK 磷酸化增加[39]。由此可知:AMPK、PGC-1α、Tfam、FOXO3 等因子可能是线粒体生物发生与线粒体未折叠蛋白反应相连通的关键因子,其中的具体机制有待于进一步证实。
2.4 线粒体融合与分裂与线粒体未折叠蛋白反应
有研究表明:UPRmt激活诱导参与线粒体分裂的基因,维持线粒体动力学稳定[6]。融合缺陷细胞的线粒体很可能含有大量不稳定的蛋白质,融合功能障碍会激活UPRmt,进而抑制由线粒体动力学异常引起的疾病,如线粒体肌病等[43]。
现有部分研究以UPRmt中的蛋白酶为切入点,通过研究蛋白酶的调控能力及泛素-蛋白酶体系统,发现了蛋白酶与线粒体的动态变化的关系:线粒体融合的几种关键效应蛋白(Mfn1、Mfn2)和分裂蛋白(Fis1、Drp1)的结构域暴露在线粒体膜的胞质侧,在线粒体分裂过程中,Drp1 和Fis1 蛋白聚集在线粒体外膜上,而Mfn1和Mfn2 蛋白被蛋白酶体泛素化和降解。在线粒体融合过程中,Mfn1 和Mfn2 蛋白水平升高,Drp1 和Fis1 蛋白定向蛋白酶体降解。这些蛋白质可与泛素-蛋白酶系统直接接触,选择性地去除融合或分裂成分,调节线粒体的动态变化[44];UPRmt中的特异性蛋白酶通过裂解OPA1直接参与调控线粒体动力学,平衡线粒体的分裂和融合[45];肌肉细胞中蛋白酶ClpP 的减少导致线粒体分裂蛋白Drp1 表达升高,即ClpP 可以通过改变线粒体动力学来保护线粒体质量[46];参与UPRmt的蛋白酶中,m-AAA 蛋白酶可诱导线粒体的分裂过程[47],i-AAA 蛋白酶参与OPA1 的加工和生成,OPA1 维持线粒体嵴结构,调控呼吸链超复杂装配过程[48]。其中,线粒体蛋白酶可将OPA1 从长OPA1(L-Opa1)切割为短OPA1(SOpa1)形式[49],长OPA1可以介导细胞中的线粒体融合,短OPA1 的表达可以促进线粒体分裂,这表明OPA1 可协调线粒体的动态变化,对于线粒体完整性和质量控制至关重要[45]。同时,OPA1 是Sirt3 的底物,可以直接去乙酰化并激活,促进线粒体融合[50]。
UPRmt的Sirt3-FOXO3a 途径可诱导Drp1、Fis1 和Mfn2 协同线粒体分裂/融合[38]。同样,在UPRmt的AKT/ERα/Omi1 转导途径中,Omi/HtrA2 蛋白酶可以调节线粒体融合蛋白Mfn2 和自噬水平,进而调控线粒体的数量和质量[51],线粒体动态变化与线粒体自噬途径协同配合,通过重新分布和去除线粒体网络中不可逆转受损的蛋白质,在应激条件下重建细胞稳态[16]。
3 运动介导线粒体未折叠蛋白反应调控线粒体质量控制
3.1 运动介导的线粒体未折叠蛋白反应调控线粒体生物发生和线粒体自噬
线粒体的生物发生和自噬的变化与运动相适应[52,53],运动导致广泛的蛋白质降解,继而进入大规模构建阶段。因此,运动可以用于蛋白质分解和重建的交叉调控[54]。有研究发现,一次运动可使骨骼肌中的Hsp70在mRNA 和蛋白水平上以强度依赖的方式升高[55],长时间的运动训练可以增加机体的Hsp70 的基线水平[56]。Hsp70 作为分子伴侣,有助于新生蛋白的正确折叠,并与非折叠蛋白相互作用,避免受损蛋白发生不适当的相互作用或降解[57]。对小鼠肌肉细胞进行慢性收缩活动,在分化4 天后骨骼肌细胞中伴侣蛋白减少,UPRmt选择性激活,Sirt3 含量升高,线粒体生物发生增强[58]。在对老年小鼠的研究中也发现有氧运动可以导致线粒体失衡,增加老年小鼠骨骼肌中Hsp60、LONP1蛋白水平[59],高强度间歇训练也对老年小鼠骨骼肌的UPRmt有明显的刺激作用[60]。这说明运动可以调控UPRmt,并引发后续的一系列反应。
在基础稳态条件下,CHOP可以通过改变电子传递链中细胞核和线粒体编码蛋白的正确化学计量来影响线粒体组成。在基础条件下,CHOP下降可导致核编码的细胞色素c 氧化酶4(cytochrome c oxidase Ⅳ,COXⅣ)表达下降,Tfam 无变化。而慢性收缩活动可以阻止由CHOP 缺乏引起的COXⅣ下降,Sirt3 和伴侣蛋白10(chaperonin 10,CPN10)升高,肌管中线粒体生物发生增强2~3 倍,说明慢性收缩活动可以通过UPRmt调控线粒体生物发生能力,改善线粒体功能。这项研究中还发现慢性收缩活动可以完全逆转因CHOP 缺失而产生的任何线粒体蛋白表达失衡,可能是由于慢性收缩活动触发了维持线粒体含量和组成的替代信号通路[58],推测慢性收缩活动触发的UPRmt可能通过其Sirt3/FOXO3A/SOD2途径或(和)AKT/ERα/OMI1途径调控线粒体的生物发生功能。同样,Hood 团队为探求在运动引起的线粒体适应过程中UPRmt起到了何种作用,对大鼠进行了不同时间跨度的慢性收缩活动,结果表明,通过PGC-1αmRNA 的线粒体生物发生信号在慢性收缩活动1 天后增加明显,继而导致细胞色素C 氧化酶活性、LC3-Ⅱ的自噬信号增加。同时,UPRmt相关伴侣蛋白Hsp70、Hsp60、10-kDa 和75-kDa 的线粒体热休克蛋白均被不同程度诱导,但慢性收缩活动诱导的线粒体适应不受CHOP 诱导的影响[58,61]。以上证据表明:慢性运动可激活线粒体UPRmt,线粒体向细胞核的逆行信号可能参与调节基因表达对运动的适应,也提示这一信号活动似乎与CHOP 信号无关,而PGC-1α可以通过抑制FOXO3介导的各种E3泛素连接酶转录的途径,抵抗肌肉萎缩进程[62]。
线粒体是细胞的能量来源,其能量产生的结果是产生活性氧(reactive oxygen species,ROS),ROS 可能导致细胞损伤、蛋白质错误折叠,最终影响机体健康或导致衰老[63]。运动这一应激的直接结果是使机体产生ROS,对于运动/骨骼肌收缩活动而言,任何运动诱导的训练适应的过程都以特定蛋白质的积累为中心,通过基因表达来促进蛋白质浓度的增加,这也是任何训练诱导反应的基础。适宜的运动类型、运动强度、运动持续时间可以通过产生少量的ROS触发UPRmt反应,产生细胞内防御机制,促进线粒体功能,而不适宜的运动则通过产生大量的ROS 触发UPRmt影响线粒体形态、含量、质量及功能,加重线粒体损伤或凋亡,进而产生机体的负向效应[64]。运动诱导的线粒体蛋白合成,部分由PGC-1α调节,在收缩活动开始时,在骨骼肌内涉及线粒体生物发生的部分早期信号开始于PGC-1α的激活[61],推测PGC-1α可能参与了UPRmt,进而改变线粒体生物发生状态[64]。同样,也有研究称ROS 参与骨骼肌中运动诱导的PGC-1α的调节[65],而ROS 是诱导UPRmt的因素之一,所以有理由相信:运动可能通过ROS 诱导的UPRmt调控PGC-1α表达,进而对线粒体的生物发生产生影响,上文提及的AMPK 也可能在此过程中扮演重要角色。
3.2 运动可能介导线粒体未折叠蛋白反应调控线粒体融合和分裂
线粒体的融合/分裂过程仅发生在应激过后的数分钟内[66],但其与有长期效应的线粒体质量控制有关[67,68],且不同的运动方式引起的线粒体融合和分裂也存在诸多不同。在生理条件下,一次性运动90 分钟后,骨骼肌中线粒体分裂标记物Fis1 和Dnm1L(编码Drp1 蛋白)以及OPA1 的表达水平升高,在运动结束后的3 小时内,这些基因的表达水平仍保持在较高水平,且Drp1 基因敲低时,肌肉细胞中的氧化代谢降低、乳酸产生增加[69],而一次性有氧运动可增加啮齿动物和人类骨骼肌中线粒体融合标志物Mfn1 和Mfn2 的mRNA 表达[70,71],但这一研究结果并未在这一领域达成共识,也有研究称一次性有氧运动不会引起线粒体融合和分裂的改变[72]。另一方面,高强度有氧运动可诱导线粒体融合蛋白Mfn1 和分裂蛋白Fis1 的蛋白表达[73],而12 周有氧运动会使人骨骼肌中Fis1 的蛋白表达量下降[74],使Mfn1和Mfn2的表达量增加[75],故不同的运动方式引起的线粒体融合与分裂的变化特征存在不一致性和不确定性。
在某些病理条件下,运动降低糖尿病患者Drp1 在Ser616位点的磷酸化水平,增加脂肪酸氧化和胰岛素敏感性[76]。同时,运动通过蛋白激酶A(protein kinase A,PKA)促进Drp1Ser637位点的磷酸化,抑制Drp1 活性,导致线粒体网络整体伸长[77,78],这种改变可能与A 型激酶锚定蛋白1(A kinase anchor protein 1,AKAP1,也称为AKAP121)的调控功能有关[79]。Hoffman 的研究也得到了相似的结论:长期耐力训练可导致高脂膳食的大鼠Ser616处p-Drp1 与总Drp1 的比值降低,Mfn2 蛋白表达升高,说明长期耐力训练可促进线粒体生物发生的平衡,表现为同时引发线粒体分裂标记物的减少和线粒体融合标记物的增加[79]。
目前虽然没有文献直接指出运动通过介导UPRmt调控线粒体融合和分裂能力,但通过前文推论可知,运动可以激活机体UPRmt,控制线粒体数量与质量。同时,UPRmt也可调控Drp1、Fis1 和Mfn2 等协同线粒体融合和分裂的主要蛋白,而运动也可以通过线粒体的融合和分裂调节线粒体质量控制机制。因此,有理由相信,运动可能通过UPRmt调控线粒体的融合与分裂,进而改善线粒体质量控制能力,但其调控机制有待于进行深入研究。
4 小结与展望
线粒体未折叠蛋白反应可以通过多种途径调控线粒体质量控制能力,使线粒体生物发生、线粒体自噬、线粒体融合与分裂等机能出现适应性改变,进而维持稳定健康的线粒体池,改善细胞生存能力,保证机体良好的机能状态。在生理状态下和应激状态下,线粒体变化的每一个过程都是相伴而生、相互依赖的,维持各过程之间的平衡尤为重要。运动作为一种特殊的应激源,可以通过介导UPRmt调控线粒体质量控制程序,维持这种平衡状态(见图1),但对上述反应的研究多集中在细胞层面,对哺乳动物中运动通过UPRmt调控线粒体质量控制的机制研究仍鲜有报道。
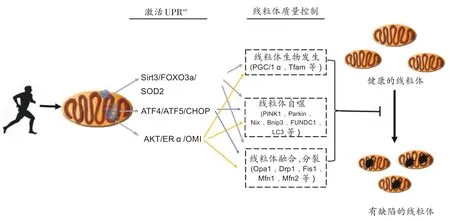
图1 运动通过线粒体未折叠蛋白反应调控线粒体质量控制机制示意图
运动可以通过介导UPRmt反应调控线粒体质量控制过程,但何种运动强度和运动持续时间是通过ROS诱发UPRmt产生线粒体正向效应的阈值?已有文献提出低氧条件可诱发UPRmt[80,81],但由UPRmt介导的最大限度促进线粒体功能的氧浓度数值范围是什么?低氧暴露主要是通过UPRmt的哪条信号转导途径发挥主要作用?各信号转导途径间是否存在交互作用?运动复合低氧暴露如何通过UPRmt影响线粒体质量控制?这些都是有待于深入研究的议题。
