H掺杂Ti3SiC2/Zr异质结的理论研究 *
2021-05-13毛彩霞胡永红陈志远
毛彩霞,胡永红,陈志远,薛 丽
(1.湖北科技学院 电子与信息工程学院,湖北 咸宁 437100;2.湖北科技学院 核技术与化学生物学院,湖北 咸宁 437100)
Ti3SiC2属于MAX相材料(Mn+1AXn,其中n = 1、2或3。M为过渡金属元素,A为主族元素,X为C或N原子)。它具有高度各向异性的六方晶体结构,既具有金属性能又具有陶瓷性能[1,2]。它还具有一系列优异的性能,如良好的高温稳定性、高导热和导电性、抗热冲击性、低密度和微观室温延展性等。这使得它广泛用于核电和航空领域的高温结构材料、化学防腐和高温加热材料[3,4]。由于具有低热中子吸收截面、合适的机械性能和良好的导热性,锆基合金是最好的水冷反应堆核材料[5,6]。但是,在富氢环境中,氢在锆基合金中的侵入和扩散很容易,这通常会导致氢脆或断裂[7]。氢脆可以通过几种方法来防止,所有这些方法的关键之处都在于尽量减少锆合金和氢之间的接触。近年来,因具有良好的抗辐射和耐腐蚀材料,Ti3SiC2被认为是锆基合金涂层材料的合适候选。最近,我们设计并研究了Ti3SiC2/Zr异质结的物理性能,重点研究了它们的力学和热学性能。研究结果表明,Ti3SiC2和Zr晶体的晶格参数匹配良好,Ti3SiC2/Zr的力学和热学性能介于Zr金属和Ti3SiC2晶体之间[8]。然而,作为良好的涂层材料,不仅要求涂层材料与主体材料的力学和热学性质匹配,更重要的是证明它能够防止腐蚀性杂质原子接触或进入主体材料[9]。因此,研究Ti3SiC2能否阻止氢通过自身从外部扩散到Zr金属中是非常重要的,这也是我们研究工作的目的。本文将氢间隙原子引入Ti3SiC2/Zr异质结,设计了24个氢原子占据不同位置的掺氢模型。经过充分的几何优化,得到了稳定的掺氢Ti3SiC2/Zr异质结体系。分析了这些掺氢Ti3SiC2/Zr异质结晶体的几何结构。计算这些掺氢模型体系的总能量,并分析Ti3SiC2/Zr异质结中氢扩散的能量随空间位置的变化。此外,还研究了掺氢对Ti3SiC2/Zr异质结电子性质的影响。我们的计算结果可为锆基合金防腐蚀涂层材料的选择提供有价值的参考。
一、计算方法和物理模型
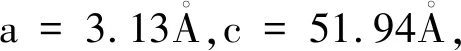
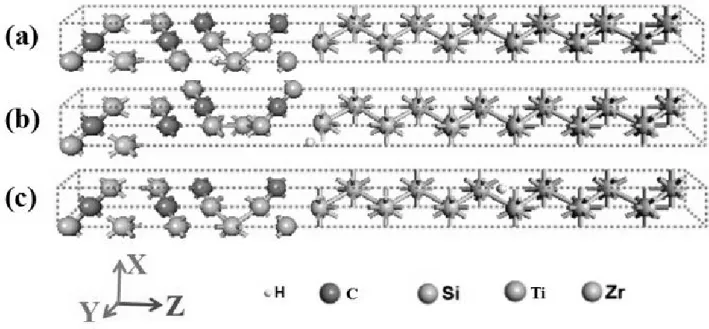
图1 三种掺杂氢原子的Ti3SiC2/Zr异质结的模型图
二、计算结果与讨论
1. 晶体结构
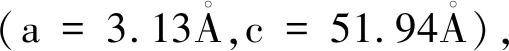
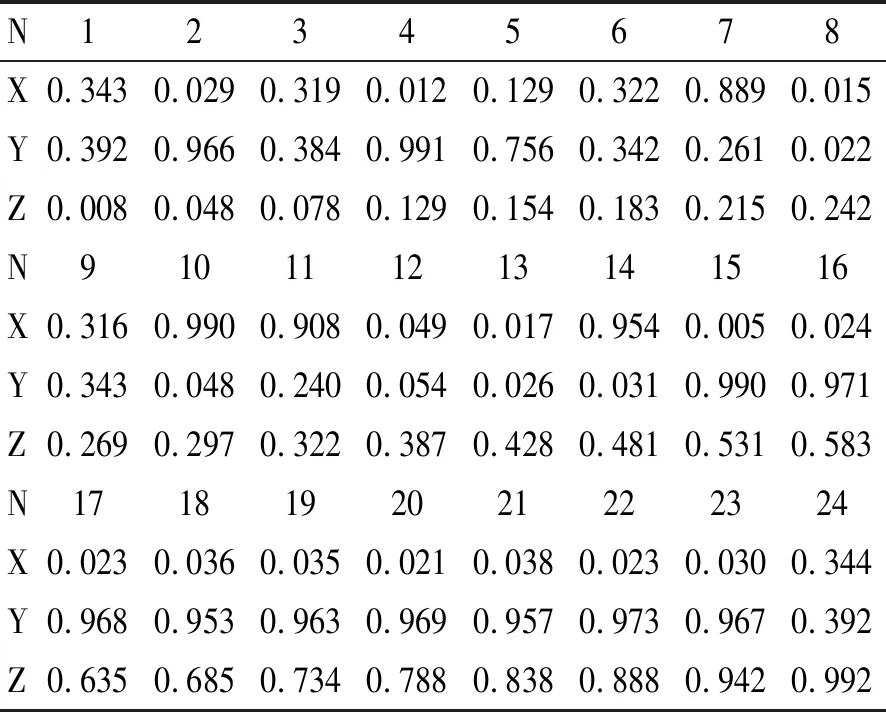
表1 24个掺杂H的Ti3SiC2/Zr超胞体系中的H原子的位置(用分数坐标X、Y、Z表示)

图2 三种掺杂氢原子的Ti3SiC2/Zr异质结的差分电子密度图
2. 能垒分析
通过几何结构优化,我们获得了24个稳定的掺H的Ti3SiC2/Zr异质结。我们在这里着重于研究总能量随Ti3SiC2/Zr超晶格中Z分数的变化(如图3所示)。从图3可以看出,间隙H原子更倾向于处在Zr金属和Ti3SiC2之间的空间中,当Z接近0.4时,体系的总能量最小。当Z从0.5变化到0.9,整个掺H的Ti3SiC2/Zr异质结系统的总能量几乎保持恒定值,比最小值高0.25 eV。当H扩散到Zr金属中时,它需要克服0.25eV的能量势垒。然而,当H原子从Zr金属向Ti3SiC2扩散时,它需要克服1.75eV的较高能垒。为什么会存在这些差异?不难理解,H原子的扩散路径会遇到不同的微观环境,即不同的原子和电子分布,并与之相互作用。这意味着间隙H原子很难从Ti3SiC2/Zr的内部异质界面处(Z=0.4)扩散到Ti3SiC2中。作为比较,我们进一步计算了H原子从真空扩散到Ti3SiC2和Zr金属时分别所遇到的势垒。我们构建了十几个掺H的Ti3SiC2/Vaccum和Zr/Vaccum超胞晶格模型,并充分弛豫。掺H的Ti3SiC2/Vaccum和Zr/Vaccum的总能量与Z轴的关系分别绘制在图4中。从图4中发现,H原子从Vaccum扩散到Ti3SiC2和Zr金属中所遇到的势垒都是正的。因此,从图3和图4的数据分析可以得出结论,Ti3SiC2是锆金属涂层材料的一种良好的候选材料,因为它可以防止H原子从真空扩散到Zr金属中。

图3 掺杂氢原子的Ti3SiC2/Zr异质结系统的总能随超胞Z分量的变化

图4 掺杂氢原子的Ti3SiC2/Vacuum和Zr/Vacuum系统的总能随超胞Z分量的变化
3.电子性质
晶体能带结构决定了材料的特性,如电子和力学性质。这里我们主要计算了三个掺H的Ti3SiC2/Zr异质结和一个未掺杂的Ti3SiC2/Zr异质结的能带结构。未掺杂的Ti3SiC2/Zr异质结的能带结构如图5所示,三个掺H的Ti3SiC2/Zr异质结的能带结构在图6中绘出。从图5和图6可以看出,所有未掺杂的Ti3SiC2/Zr和掺H的Ti3SiC2/Zr异质结的能带结构图都出现了跨越费米能级的导带,这表明所有这些系统都具有金属性质。很明显,掺H并不改变Ti3SiC2/Zr异质结的金属性质,但改变了它们的能带结构(如图5和6所示)。
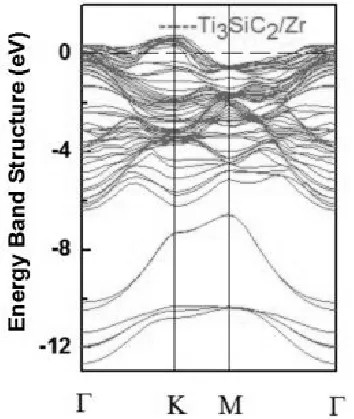
图5 未掺杂的Ti3SiC2/Zr异质结的电子能带结构图
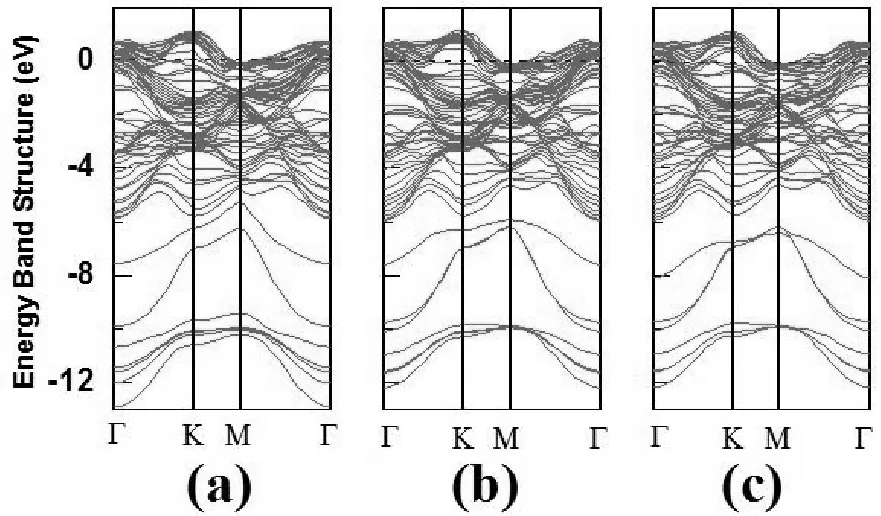
图6 三种掺杂氢原子的Ti3SiC2/Zr异质结的电子能带结构图
未掺杂的Ti3SiC2/Zr异质结和掺H的Ti3SiC2/Zr异质结的总电子态密度(TDOS)和偏态电子密度(PDOS)用GGA-PBE函数计算。计算结果分别绘制成图7和图8。从图7和图8中,我们可以看到N (EF)值都大于零,这意味着这些未掺杂的Ti3SiC2/Zr异质结和掺H的Ti3SiC2/Zr异质结是金属性的。氢的引入对Ti3SiC2/Zr异质结的扩散系数影响很小,这与能带结构的变化一致。为了进一步解释Ti3SiC2/Zr异质结中氢原子的引入所引起的DOS的微小变化,我们详细研究了各种轨道的杂化情况。从图7和图8所示的Ti3SiC2/Zr的TDOS,观察到八个主峰,即价带中的PI、PII、PIII、PIV、PV、PVI、PVII,和导带中的PⅧ,它们由不同的原子的不同轨道贡献产生。在DOS图中,大多数峰是由不同的原子分轨道贡献的,不同轨道的杂化反映了未掺杂和掺H的Ti3SiC2/Zr异质结的共价性本质特性。此外,在图8中发现掺H影响了Ti3SiC2/Zr异质结DOS的PⅠ、PⅢ和PⅦ峰。
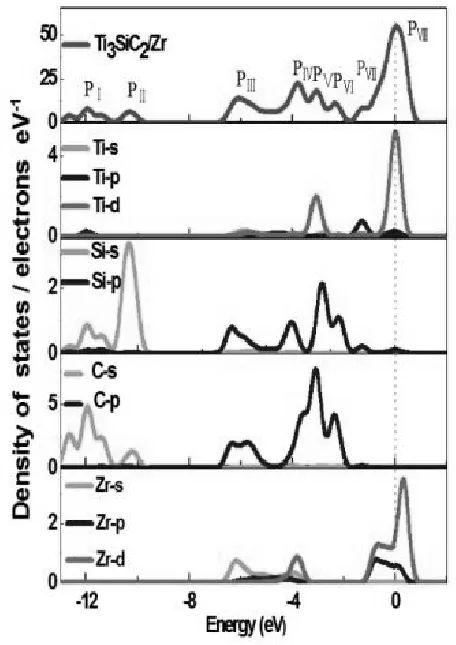
图7 未掺杂的Ti3SiC2/Zr异质结的总(分)电子态密度图
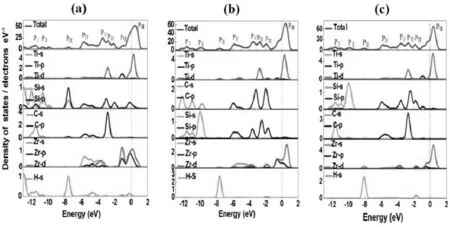
图8 三种掺杂氢原子的Ti3SiC2/Zr异质结的总(分)电子态密度图
在模型(a)的情况下,H原子的1s轨道与C原子的2s轨道、Si原子的3s轨道和3p轨道杂化。C-2s与H-1s不形成共价键,Si-3sp与H-1s也不形成共价键,这表现在掺H的Ti3SiC2/Zr异质结的总电荷等值线图中(如图9所示)。

图9 掺杂氢原子的Ti3SiC2/Zr异质结的总电荷密度等值线图
在模型(b)中,H原子的1s轨道只与Ti原子的3p轨道杂化。在模型(c)的情况下,H原子的1s轨道与Zr原子的5s、4p和4d轨道以及Si原子的3p轨道杂化。在这三种模型中,间隙H原子与周围原子的杂化都很弱,因此,Ti3SiC2/Zr的键性质略有变化,掺H的Ti3SiC2/Zr的能带结构与未掺杂Ti3SiC2/Zr材料有很小的差异。我们还计算了掺H的Ti3SiC2/Zr的Mulliken电荷分布。一般来说,Ti原子总是由于电子丢失而带正电荷。H、Si和C原子得到电子,所以它们是负电荷。由于表面的Zr原子向Ti3SiC2转移了少量的电子,所以是正电荷。计算结果表明,Ti3SiC2/Zr中从Ti原子到Si原子和C原子的电荷转移总数为3.05。这意味着Ti-Si键和Si-C键具有离子键性质(如图9所示)。此外,还观察到相同数量的电荷(0.02)从Ti3SiC2材料转移到Zr系统。总的来说,能带结构、电子态密度和Mulliken电荷分布分析表明,掺H的Ti3SiC2/Zr异质结都具有金属、共价和离子性质。
三、结语
本文用第一性原理方法分析了掺H的Ti3SiC2/Zr异质结的几何结构、能垒和电子性质。首先,设计并优化了24种掺H的Ti3SiC2/Zr异质结晶体的几何结构,发现H原子作为间隙原子处在Ti3SiC2/Zr异质结中,不影响其晶格结构。其次,计算了24个掺H的Ti3SiC2/Zr异质结的总能量,分析了Ti3SiC2/Zr异质结中H扩散的能垒。发现H在Zr金属中容易扩散,因为H遇到的能垒相对较低(约0.25电子伏)。然而,H原子从外部扩散到Ti3SiC2中的能垒很高(1.75电子伏),这意味着Ti3SiC2是一种很有前途的防止核工业中Zr合金氢脆和腐蚀的涂层材料。最后,研究了掺H的Ti3SiC2/Zr异质结的能带结构、电子态密度和Mulliken电荷分布。计算结果表明,这些材料仍然具有金属、共价和离子性质,H原子的引入对其电子性质影响不大。
