IL‐6/STAT3信号通路在结直肠癌发展和治疗中的进展
2021-05-11何子勤刘子钰佘晓敏李双航卢露叶甲舟梁嵘林燕
何子勤 刘子钰 佘晓敏 李双航 卢露 叶甲舟 梁嵘 林燕
结直肠癌(colorectal cancer,CRC)是全球十大恶性肿瘤之一,2018年全球新发CRC 180多万例,约占癌症病例的1/10,死亡率高居全球第二位[1]。在我国,CRC发病率逐年增长,2018年新发CRC 52.1万例,发病率位居所有恶性肿瘤的第二位[2]。因此,探究CRC的发展机制及治疗方法尤为重要。CRC发病机制繁多复杂,与肠道菌群紊乱、免疫反应异常、遗传等有关,其中肠道炎症是最重要的因素之一[3]。研究表明,肠道炎症向CRC发展经历了众多信号通路紊乱,其中IL⁃6/STAT3通路可直接促进肿瘤细胞增殖和存活,被认为是CRC发生的关键通路之一[4]。本文就IL⁃6/STAT3信号通路在介导CRC发展及其在CRC治疗的研究进展进行阐述。
1 IL‐6/STAT3信号通路
IL⁃6于1986年首次被成功克隆,作为一种多效细胞因子,在免疫调节、造血、炎症和肿瘤发生方面具有重要作用[5]。研究表明,IL⁃6在CRC组织中表达显著增加,并与肿瘤大小、分期和生存率密切相关,其介导的信号传导也与CRC发展密切相关[6]。IL⁃6与受体形成复合物从而激活下游信号通路,其受体由2个亚基构成:分子大小为80 kDa的配体结合链IL⁃6R(IL⁃6Ra,CD126)和分子量为130 kDa的信号传导链糖蛋白 130(glycoprotein130,gp130)[7]。IL⁃6 信号传导途径分为经典信号通路和反式信号通路。在经典信号传导途径中,胞外IL⁃6与膜上的IL⁃6R(membrane⁃bound IL⁃6R,mIL⁃6R)结合形成复合物,诱导gp130聚集并与之结合,形成了由2个IL⁃6,2个IL⁃6R及2个gp130分子构成的异六聚体,随后复合体激活Janus激酶(Janus kinase,JAK),后者通过激活 STAT3形成二聚体进入细胞核调控靶基因转录[8]。IL⁃6的反式信号传导途径与经典信号传导途径基本相同,不同之处在于反式信号通路中与IL⁃6结合的受体是可溶性IL⁃6R(soluble IL⁃6R,sIL⁃6R)而非mIL⁃6R[9]。sIL⁃6R由mIL⁃6R的有限蛋白水解或IL⁃6R mRNA可变剪切产生[10⁃11]。经典信号通路与反式信号通路表达范围并不相同。与gp130在所有细胞中表达不同[12],mIL⁃6R主要表达于中性粒细胞、单核细胞、活化的B细胞、CD4+和肝细胞,因此IL⁃6经典信号途径表达的范围有限[13];而IL⁃6与血清中sIL⁃6R形成的复合物,可以作用于所有表达gp130的细胞,因此反式信号通路扩大了IL⁃6/STAT3信号通路的作用范围[14]。在功能上,IL⁃6经典信号通路主要诱导急性期反应,具有抗炎作用;与之相反,反式信号传导与促炎反应有关[15]。目前认为,IL⁃6对肿瘤的刺激主要由IL⁃6反式信号途径介导(图1)[16]。
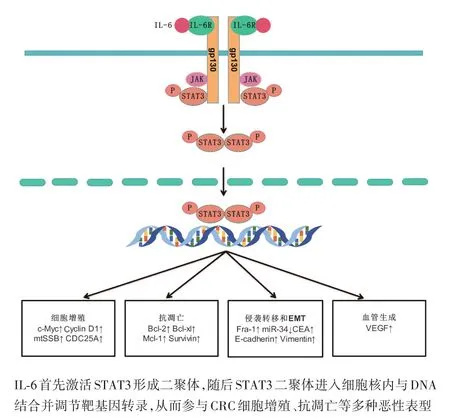
图1 IL‐6/STAT3信号通路促进CRC发展的调控途径
2 IL‐6/STAT3信号通路促进CRC发展
IL⁃6信号介导STAT3持续激活对CRC增殖、抗凋亡、侵袭转移等多种恶性行为有重要的驱动作用。
2.1 细胞增殖
Myc是一种原癌基因,可调控下游靶基因转录,参与细胞增殖、分化等细胞过程[17]。目前发现多数CRC患者存在c⁃Myc蛋白过表达,部分患者还出现c⁃Myc基因扩增,提示病情恶化和预后欠佳[18]。在CRC中,IL⁃6激活STAT3,介导c⁃Myc快速激活并引发代谢紊乱和肿瘤进展[19]。KIUCHI等发现STAT3在IL⁃6刺激下介导c⁃Myc激活,而STAT3能与c⁃Myc P2启动子中的E2F位点(98TTGGCGGGAAA105)结合,而对该位点进行点突变,发现其对IL⁃6的响应下降,c⁃Myc表达也随之减少[20]。
在CRC中IL⁃6/STAT3通路还能促进细胞周期蛋白D1(Cyclin D1)表达。Cyclin D1可驱动细胞周期从G1期到S期转变,细胞核内积累的Cyclin D1可促进细胞增殖[21⁃22]。LIU等[23]利用茶多糖抑制小鼠CRC细胞中的IL⁃6/STAT3通路,发现下游Cyclin D1表达降低。此外,还有研究发现Cyclin D1启动子序列中存在STAT3结合位点(也称GAS位点),破坏此位点可致STAT3介导的转录激活减少[24]。
LNRRIL6(long noncoding RNA regulating IL⁃6 transcription)是新鉴定的lncRNA,在CRC中上调并促进CRC细胞增殖;还可直接与IL⁃6启动子结合并介导其表达上调,增强STAT3激活,CDC25A、Cyclin D1也随之显著上调[25]。
IL⁃6/STAT3信号通路还可通过诱导细胞中线粒体单链DAN结合蛋白(mitochondrial single⁃strand DNA binding protein,mtSSB)的表达而介导端粒酶激活从而促进CRC细胞增殖。IL⁃6/STAT3信号通路诱导其下游转录因子FOXP1激活,随后FOXP1与mtSSB基因启动子的关键转录调节区(−800~−700之间的100 bp区域)结合,从而促进其表达。过表达的mtSSB激活ROS/Akt/mTOR通路促进人端粒酶逆转录酶表达,增强CRC细胞端粒酶活性及稳定端粒,从而促进CRC增殖[26]。
2.2 抗凋亡
IL⁃6反式信号通路不仅能调控细胞增殖,也是肿瘤细胞抵抗凋亡的关键通路。阿司匹林是一种能够抑制IL⁃6表达的药物。TIAN等采用阿司匹林治疗CRC小鼠,发现阿司匹林治疗组小鼠体内IL⁃6表达明显较低,磷酸化STAT3的水平同样减少,STAT3的下游靶基因Bcl⁃2、Bcl⁃xl表达显著降低,CRC小鼠体内肿瘤细胞凋亡显著增加[27]。还有研究报道,IL⁃6/JAK/STAT3信号通路可上调靶基因Mcl⁃1,保护CRC细胞免受肿瘤坏死因子相关凋亡配体诱导的细胞凋亡[28]。STAT3通过与Mcl⁃1中的SIE元件结合并促进其表达[29]。此外,IL⁃6/STAT3激活的抗凋亡靶基因还有Survivin[23]。Survivin启动子上存在STAT3的反应元件,IL⁃6介导激活的STAT3能通过与Survivin启动子的结合并调控其转录表达上调[30⁃31]。
2.3 侵袭转移和上皮间质转化
IL⁃6/STAT3信号通路还能增强CRC的侵袭转移能力,其机制可能与fos相关抗原⁃1(fos like antigen 1,Fra⁃1)有关。而Fra⁃1已被证实在肠癌细胞转移扩散中起至关重要的作用[32]。研究发现IL⁃6介导激活的STAT3,经K685乙酰化和Y705磷酸化后可直接与位于转录起始位点上游−600 bp附近的Fra⁃1启动子结合,并上调其表达。随后,Fra⁃1通过上皮间质转化诱导转录因子和基质金属蛋白酶增强CRC细胞侵袭能力[33]。
microRNA⁃34a(miR⁃34a)由肿瘤抑制因子P53诱导[34],可下调Snail1表达而抑制上皮间质转化并阻遏癌症发展[35]。在CRC中,STAT3可结合至miR⁃34a第一个内含子中的结合位点从而抑制其表达,进而使Snail1上调。而IL⁃6R也是miR⁃34a的标靶之一,miR⁃34a可通过IL⁃6R的结合位点抑制其表达,因此过表达的IL⁃6可介导STAT3对miR⁃34a的抑制,反过来也促进了IL⁃6R的表达,激活IL⁃6/STAT3信号通路。同时,IL⁃6R/STAT3/miR⁃34a反馈环的激活也维持了对miR⁃34a的抑制,从而促进CRC上皮间质转化和侵袭转移[36⁃37]。IL⁃6/STAT3 信号通路还能通过降低E⁃钙黏着蛋白(E⁃cadherin)表达并上调波形蛋白(vimentin),从而增强CRC细胞的上皮间质转化而促进细胞迁移和侵袭[38]。
此外,癌胚抗原因子(carcinoembryonic antigen,CEA)作为CRC最主要的肿瘤标志物,对CRC细胞侵袭和转移也具有促进作用[39⁃40]。研究发现,采用Hyper⁃IL⁃6(介导 IL⁃6反式信号)处理CRC细胞后,激活的 STAT3能够上调 HIF⁃1α,HIF⁃1α随之通过与CEA启动子上的正调控元件EP⁃1结合,进一步促进 CEA 的表达[41⁃43]。
2.4 血管生成
IL⁃6/STAT3信号转导通路刺激肿瘤血管生成与血管生成因子(vascular endothelial growth factor,VEGF)激活密切相关。VEGF作为最直接有效的血管生成因子,已被证明在CRC中呈高表达,且与预后呈负相关[44]。研究表明CRC患者中IL⁃6和VEGF水平明显高于健康对照组,并与疾病进展相关[45]。XIONG等[46]沉默STAT3后发现,CRC细胞中VEGF分泌减少,提示STAT3能够调控VEGF的表达。NIU等[47]进一步证实人VEGF启动子中存在STAT3结合位点,激活的STAT3突变体(STAT3C)能够直接与VEGF启动子结合,促进VEGF转录和肿瘤血管生成,而该位点发生突变后,STAT3C诱导VEGF启动子的活性也随之消失。
2.5 IL⁃6/STAT3信号通路与肠道菌群
结直肠寄居着复杂的微生物群落,其中以细菌数量最多,称为肠道菌群。肠道菌群与肠道上皮细胞相互作用,参与消化吸收、免疫调节等过程,具有增强肠道屏障、维持肠道稳态等重要作用[48]。当肠道菌群的结构、数量和种类发生改变,即肠道菌群失调时,可通过引起肠道免疫调节失衡和致癌物质增加等方式促进CRC发生发展[49]。研究表明,CRC患者肠道菌群在个体丰度上具有显著差异[50],且可通过多种复杂的机制直接或间接促进CRC发生发展,如毒力因子的生成、肠道代谢产物的改变等[51]。肠道菌群失调可引起慢性结直肠炎导致的组织损伤,继发氧化应激造成肠上皮细胞DNA损伤,诱发不典型增生并促进癌变。此外,肠道菌群失调构建的微环境还有助于募集各种炎症细胞,通过分泌释放包括IL⁃6、IL⁃8等炎症因子,激活STAT3等信号通路而抑制肿瘤免疫反应[52]。
3 IL‐6/STAT3信号通路抑制剂在CRC治疗中的临床研究
目前,不少针对CRC IL⁃6/STAT3信号通路的靶向药物的临床试验正在开展,如IL⁃6抑制剂(siltux⁃imab),JAK抑制剂(itacitinib),STAT3抑制剂(OPB⁃31121,AZD9150 及 TTI⁃101)[53⁃58](表 1)。
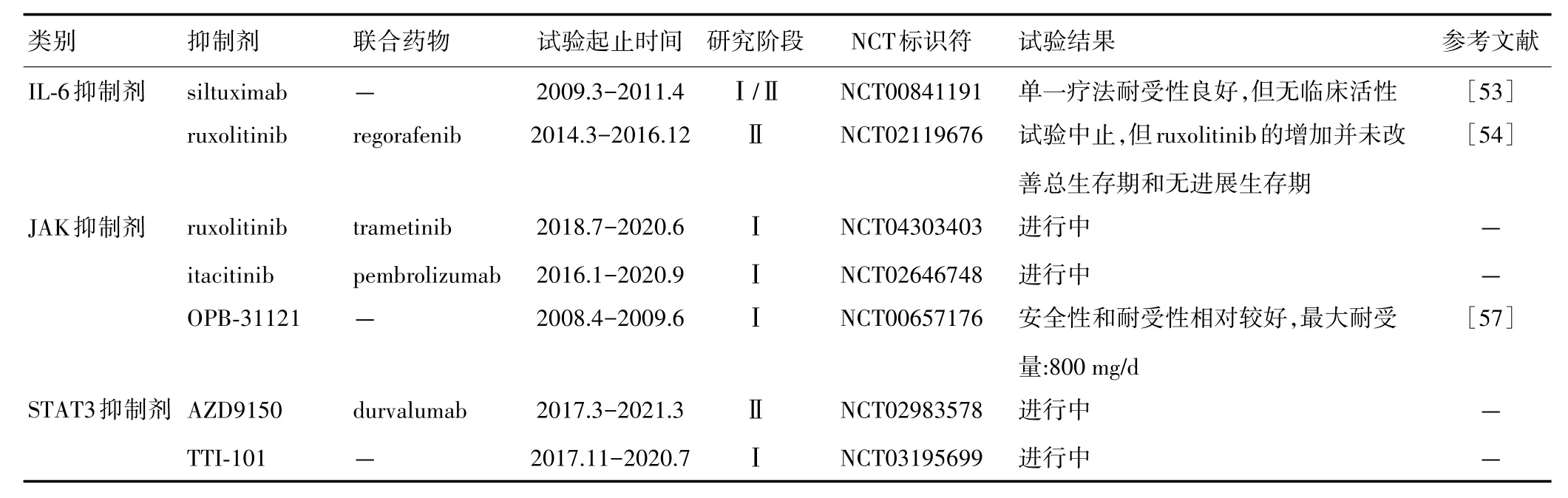
表1 靶向CRC IL‐6/STAT3信号通路的临床试验
siltuximab是针对IL⁃6的单克隆抗体,也是目前FDA批准用于治疗多中心型卡斯特曼病的唯一药物。但siltuximab单一疗法在包括CRC在内的晚期实体瘤患者Ⅰ/Ⅱ临床试验中并未显示出临床活性。研究结果表明,纳入试验的35例CRC患者疾病并没有得到有效缓解,siltuximab对晚期CRC患者临床获益有限[53]。
一项临床试验将JAK1/JAK2的选择性抑制剂ruxolitinib联合regorafenib用于治疗难治性转移CRC。但纳入试验的396例患者的总生存期和无复发生存期均未显著改善,表明ruxolitinib联合regorafenib并未提高转移CRC患者的疗效[54]。一项ruxolitinib联合trametinib治疗RAS突变型CRC的临床试验也正在进行中(NCT04303403),结果值得期待。itacitinib是一种新型口服JAK1选择性抑制剂[55]。itacitinib联合pembrolizumab治疗CRC患者的Ⅰ期试验近期已结束,但尚无结果发布(NCT02646748)。
STAT3抑制剂研发是学者关注的焦点之一。OPB⁃31121与STAT3的SH2结构域具有高亲和力,是新型的STAT3抑制剂,具有明显抗癌活性[56]。Ⅰ期临床研究显示,OPB⁃31121对晚期CRC患者具有良好的抗肿瘤活性,最大耐受剂量为800 mg/d[57]。作为STAT3反义寡核苷酸,AZD9150能够促进STAT3 mRNA破坏或抑制其翻译,是唯一进入临床试验的STAT3反义分子[58]。MD安德森癌症中心于2017年3月发起了一项Ⅱ期临床试验(NCT02983578):AZD9150联合抗PD⁃L1抗体durvalumab治疗CRC患者,预计2021年3月结束。TTI⁃101是由Tvardi Therapeutics公司开发的STAT3口服抑制剂。目前已经开展TTI⁃101治疗包括CRC在内的晚期实体瘤的Ⅰ期试验(NCT03195699)。新型STAT3抑制剂 Bruceantinol(BOL)能够强烈抑制STAT3结合DNA的能力。在体内和体外CRC模型中,BOL均显示出显著的抗癌活性,但其临床试验有待开展[59]。截至目前,尚无任何靶向IL⁃6/STAT3信号通路的抑制剂被批准用于CRC治疗,因此有待进一步研发针对CRC IL⁃6/STAT3信号通路靶向药物的临床应用价值。
4 小结
在当前全球CRC疾病负担不断加重情况下,探究CRC发生发展的作用机制对预防、诊断和治疗CRC尤为重要。IL⁃6/STAT3信号通路作为CRC发展的关键通路之一,通过介导众多下游致癌靶基因的激活促进CRC的发生发展。近年来,临床数据还表明靶向IL⁃6/STAT3信号通路对CRC治疗有益,针对该通路及其下游信号通路的药物研究也是未来治疗CRC的研究热点之一。然而鉴于IL⁃6/STAT3信号通路促进CRC发展机制的复杂性,仍有一些致癌机制尚未明确,这也影响了治疗CRC药物研发进程,因此仍需进一步研究IL⁃6/STAT3信号通路促进CRC发展的作用机制及其靶向药物。
