LncRNA ZEB1‐AS1对B细胞非霍奇金淋巴瘤增殖和凋亡的影响及其下游调控网络的分析
2021-05-11廖成成邓木英莫国君雷丹青夏爱军荣超谭晓虹岑洪卢盛娟
廖成成 邓木英 莫国君 雷丹青 夏爱军 荣超 谭晓虹 岑洪 卢盛娟
B细胞非霍奇金淋巴瘤(B⁃cell non⁃Hodgkin's lymphoma,B⁃NHL)是一种淋巴造血系统恶性肿瘤,其中弥漫大B细胞淋巴瘤(diffuse large B⁃cell lymphoma,DLBCL)是最常见的类型,占非霍奇金淋巴瘤的30%~58%[1]。化疗和免疫化疗是B⁃NHL常用的治疗手段,但部分患者一线治疗后耐药或复发难治,疗效并不理想[2⁃3]。目前在临床实践中,淋巴瘤一线方案均以蒽环类药物为主,如多柔比星(doxorubicin,Dox)。因此,探讨Dox耐药机制,寻找B⁃NHL诊断和治疗新型生物标志物及治疗靶点尤为重要。
淋巴瘤发生发展与转录异常有关,除mRNA水平异常外,还包括基因组中ncRNAs调控能力异常(lncRNAs、miRNAs等)[4]。近年来SALMENA等提出细胞内有一类竞争性内源性RNA(competing endoge⁃nous RNA,ceRNA)假说[5],认为 lncRNAs可通过miRNA反应原件(miRNA response elements,MREs)吸附miRNA,抑制miRNA作用,间接调控蛋白编码基因的表达,从而调节肿瘤进程。可见miRNA在该调控网络中起关键作用[6⁃7]。本研究通过GEO数据集预测与DLBCL预后相关的lncRNAs,探讨lncRNA ZEB1⁃AS1及其与Dox协同对B⁃NHL细胞增殖和凋亡的影响,并构建lncRNA⁃miRNA⁃mRNA ceRNA调控网络,预测下游核心基因,为B⁃NHL诊断和治疗提供新的靶点。
1 材料与方法
1.1 数据来源及分析
数据集1来源于GEO数据集(GSE31312)中DLBCL表达谱数据及患者样本信息(498例)。纳入标准:⑴病理确诊为DLBCL;⑵可获得患者的总体生存时间;⑶可获得标准化lncRNA及mRNA的表达数据。表达谱数据用R语言limma包进行差异基因的筛选;构建Cox回归模型,按照HR>1.5,调整后的P值<0.05,挑选出前20个高风险lncRNA。数据集2为从UCSC XENA数据库(https://xenabrowser.net/datapages/)[8]中选取444例正常组织与47例DLBCL组织的表达数据集,比较HR较高的前8个基因在正常组织和肿瘤组织中的表达量。通过文献查阅,筛选出在数据集1中HR较高同时在数据集2肿瘤组织中显著高表达,且未被文献报道过的lncRNAs,并采用qPCR验证其在正常组织(外周血单个核细胞)和B⁃NHL细胞系中的表达量,最终确定lncRNA ZEB1⁃AS1为本研究的目的lncRNA。
根据ZEB1⁃AS1表达的中位数将数据集1中的患者分为高、低表达组,运用limma数据包对数据集进行mRNA的共表达分析,若表达趋势一致则定义为共表达基因。同时利用DIANA tools查询经实验验证的编码和非编码RNA上的miRNA靶标数据库(TarBase v7.0和LncBase)。通过DIANA⁃miRPath(http://diana.imis.athena⁃innovation.gr/)算法预测与lncRNA ZEB1⁃AS1相结合的miRNA及其结合位点,并运用Targetscan网站(http://www.targetscan.org/vert_71/)预测miRNA的靶基因。将与lncRNA ZEB1⁃AS1共表达的mRNA与Targetscan预测的靶基因取交集得到ceRNA调控网络下游基因,再将lncRNA⁃miRNA⁃mRNA调控轴导入Cytoscape 3.6.0并进行可视化输出,接着采用R语言的clusterProfiler包对差异表达基因进行基因本体GO功能富集分析和KEGG信号通路分析,最后利用STRING数据库(https://string⁃db.org/)对差异基因的编码蛋白进行分析并构建蛋白相互作用网络(PPI)。
1.2 细胞系及主要试剂
B⁃NHL细胞系Raji、Ramos、Farage和293T购自上海富衡生物科技有限公司。外周血单个核细胞(peripheral blood mononuclear cell,PBMC)为健康供者外周血经梯度离心获得;Gibco胎牛血清购自美国Life公司;DMEM高糖培养基、RPMI 1640培养基购自美国Hyclone公司;慢病毒载体SV40及目的质粒sh⁃NC、sh⁃ZEB1⁃AS1购自VectorBuilder Inc,美国芝加哥;慢病毒包装质粒PMD2.G、PSPAX2购自Addgene公司;Lipofectamine 3000购自美国Thermofisher公司,Polybrene购自美国Sigma公司,Dox购自深圳万乐药业有限公司;CCK⁃8试剂盒购自江苏凯基生物技术股份有限公司;Annexin V⁃FITC/PI凋亡检测试剂盒购自美国BD公司;TRIzol试剂盒、逆转录试剂盒和SYBR RT⁃qPCR试剂盒购自TaKaRa公司。
1.3 细胞转染及分组
将Raji和Ramos细胞接种至含10%胎牛血清的RPMI 1640培养基,293T细胞接种至含10%胎牛血清DMEM培养基,并置于37℃、5% CO2、95%湿度培养箱中培养。按照试剂盒说明书的操作,使用Lipofectamine 3000在293T细胞转染目的质粒生产病毒后,予Polybrene辅助感染Raji或Ramos细胞,构建 ZEB1⁃AS1敲降细胞。采用RT⁃qPCR检测转染情况,嘌呤霉素筛选稳定转染细胞株,然后以不同浓度(50 ng/mL、100 ng/mL、200 ng/mL、400 ng/mL)Dox处理 Raji和Ramos细胞,设未经处理的相应对照组。实验分为4组:⑴慢病毒对照组:sh⁃NC+DMSO;⑵ZEB1⁃AS1敲低组:sh⁃ZEB1⁃AS1+DMSO;⑶Dox组:sh⁃NC+Dox;⑷ZEB1⁃AS1敲低+Dox组:sh⁃ZEB1⁃AS1+Dox。
1.4 RT⁃qPCR法检测B⁃NHL细胞中ZEB1⁃AS1 mRNA的表达情况
采用Trizol提取法提取各组细胞的总RNA。根据逆转录试剂盒说明书,分别取各组细胞总RNA 1 μg进行逆转录合成cDNA,再按照定量PCR试剂盒说明书,以cDNA为模板进行PCR反应。PCR反应条件:95℃、15s;60℃、1min,40个循环。引物序列:ZEB1⁃AS1的上游引物为 5'⁃GTGGGCACTGCTGAATTTGA⁃3',下游引物为5'⁃GCGGAACTTCTAGCCTCTCT⁃3';GAPDH的上游引物为5'⁃GCACCGTCAAGGCTGAGAAC⁃3',下游引物为5'⁃TGGTGAAGACGCCAGTGGA⁃3'。实验重复3次。以GAPDH作为内参,使用2−△△Ct法进行相对定量的数据分析。
1.5 CCK⁃8法检测B⁃NHL细胞的增殖活力
取稳定转染且经不同浓度Dox处理后的Raji和Ramos细胞接种到96孔板(4×103/孔)中培养,每组设3个复孔。培养48 h后,向各孔分别加入20 μL CCK⁃8溶液,恒温孵育2 h,于酶标仪检测各孔在450 nm波长处的吸光度(A)。实验重复3次。细胞增殖活力(%)=[(实验组A值−对照组A值)/(空白组A值−对照组A值)]×100%。
1.6 Annexin V⁃FITC/PI流式细胞术检测B⁃NHL细胞的凋亡
取稳定转染且经Dox(0 ng/mL和100 ng/mL)处理后的Raji和Ramos细胞接种到24孔板(1×105/孔)中,置于5% CO2、37℃、饱和湿度的恒温培养箱培养48 h。收集细胞培养液于1.5 mL离心管,并洗涤2次,取500 μL Binding Buffer重悬细胞。按照 Annexin V⁃FITC/PI凋亡检测试剂盒的操作步骤,每管依次加入5 μL Annexin V⁃FITC和5 μL PI,充分混匀,室温避光孵育15 min,应用流式细胞仪检测细胞凋亡率。实验重复3次。
1.7 统计学方法
采用R语言(version 3.6.0)对数据进行统计分析。差异表达基因的筛选标准均为:截断值logFC>1.5,Bonferroni法调整后的P值<0.05[9],并对差异基因进行标准化处理。多组均数比较采用单因素方差分析,若组间差异有统计学意义,则需进行多重比较。多个实验组与对照组比较的多重比较采用Dunnett,两两比较采用Tukey检验。两组均数比较采用独立样本t检验。采用R软件的survival软件包应用Kaplan⁃Meier法计算总生存率并绘制生存曲线,组间差异比较采用log⁃rank检验法。以P<0.05表示差异具有统计学意义。
2 结果
2.1 目标lncRNAs的筛选
通过GEO数据集(GSE31312)筛选前20个可预测 DLBCL 不良预后的高风险 lncRNAs(HR:4.46~15.56),见图1A。采用UCSC XENA数据库对RNAseq数据进行log2转化后的正常组织与DLBCL组织的lncRNAs表达量进行比较,8个高风险lncRNAs中,除SPATA13(P≥0.05)外,ZFAS1、NNT⁃AS1、LINC01578、RPARP⁃AS1、ZEB1⁃AS1、EBLN3P、OTUD6B⁃AS1等在DLBCL组织中的表达均高于正常组织(均P<0.01),见图1B。RT⁃qPCR检测结果显示,ZEB1⁃AS1在淋巴瘤细胞系Raji、Ramos、Farage的表达倍数分别是PBMC的(4.67±0.21)倍、(3.13±0.15)倍、(5.33±0.25)倍(均P<0.05),见图1C。GSE31312数据集分析显示,ZEB1⁃AS1高表达组DLBCL患者(249例)中位生存时间低于低表达组(249例)(45个月vs57个月,P<0.001),见图1D。选择ZEB1⁃AS1进行后续实验。
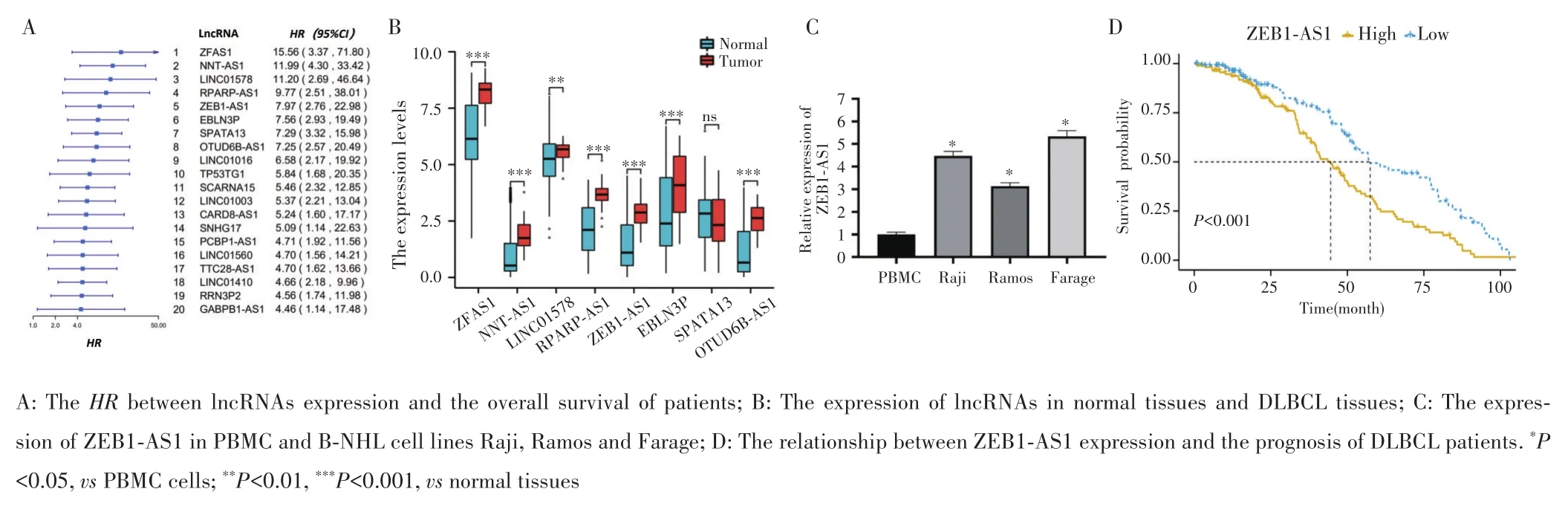
图1 目标lncRNAs的筛选Fig.1 Screening of target lncRNAs
2.2慢病毒感染B⁃NHL细胞后ZEB1⁃AS1的表达情况
RT⁃qPCR 检测结果显示,与 sh⁃NC 组相比,sh⁃ZEB1⁃AS1#1组和sh⁃ZEB1⁃AS1#2组细胞中 ZEB1⁃AS1表达量均降低(均P<0.05),其中 sh⁃ZEB1⁃AS1#1敲降效果最好,见图2。表明成功构建ZEB1⁃AS1敲低的 B⁃NHL细胞,用sh⁃ZEB1⁃AS1#1组细胞进行后续实验。
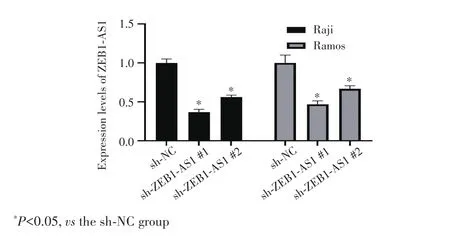
图2 RT‐qPCR检测慢病毒感染B‐NHL细胞后ZEB1‐AS1的表达情况Fig.2 The expression of sh‐ZEB1‐AS1 in B‐NHL cells after lentivi‐rus infection detected by RT‐qPCR
2.3 ZEB1⁃AS1敲低及联合Dox对B⁃NHL细胞增殖活力的影响
CCK⁃8检测结果显示,敲低ZEB1⁃AS1后,Raji和Ramos 细 胞增殖活力均较相应对照组降低(P< 0.05);与不同浓度(50 ng/mL、100 ng/mL、200 ng/mL、400 ng/mL)Dox联合处理后,细胞增殖活力明显受抑制(均P<0.05),且细胞杀伤作用呈浓度依赖性,其中100 ng/mL和50 ng/mL浓度Dox作用下细胞活性抑制作用最大,协同分数分别为15.008和13.249,见图3。

图3 ZEB1‐AS1敲低及联合不同浓度多柔比星对Raji和Ramos细胞活力的影响Fig.3 The effect of ZEB1‐AS1 knockdown and combined with different concentrations of doxorubicin on the viability of Raji and Ramos cells
2.4 ZEB1⁃AS1敲低及联合Dox对B⁃NHL细胞凋亡的影响
流式细胞术检测结果显示,与sh⁃NC+DMSO组相比,sh⁃ZEB1⁃AS1+DMSO组、sh⁃NC+Dox组、sh⁃ZEB1⁃AS1+Dox组细胞凋亡率均显著升高(均P<0.05),且sh⁃ZEB1⁃AS1+Dox 组细胞凋亡率明显高于sh⁃NC+Dox组(P<0.05),见图4。
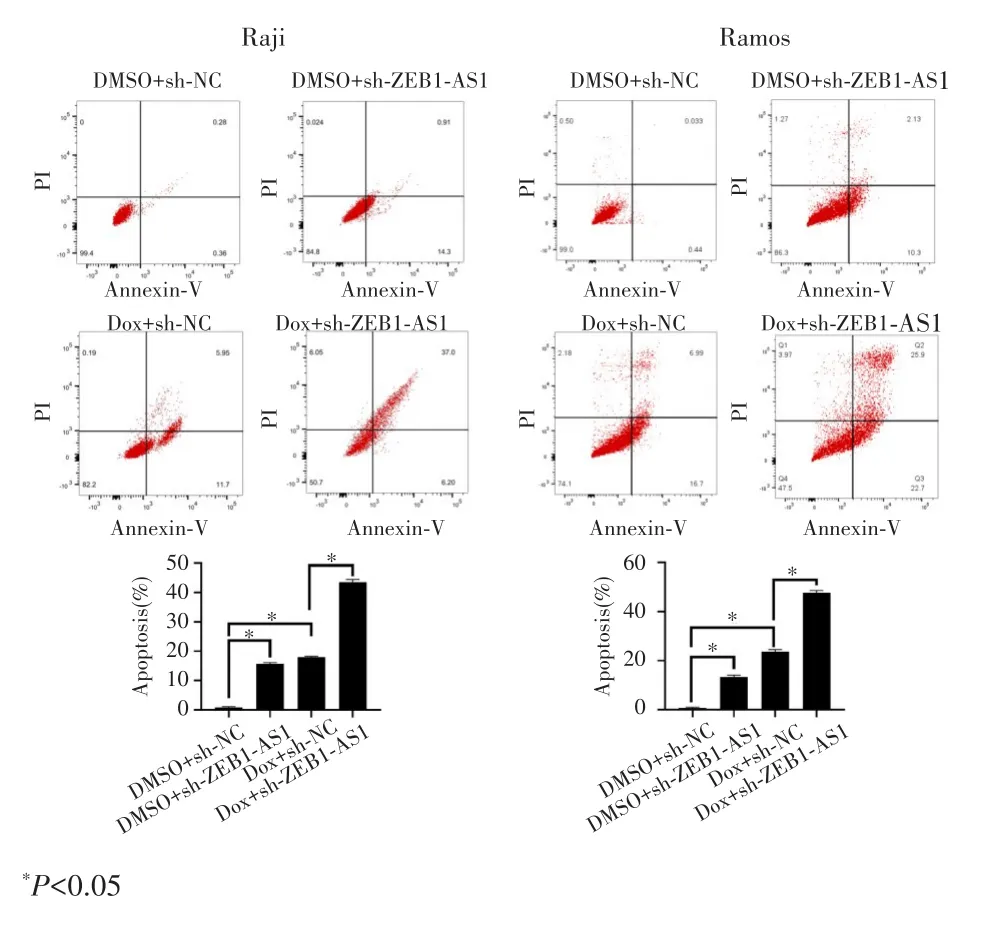
图4 ZEB1‐AS1敲低及联合多柔比星对Raji和Ramos细胞凋亡的影响Fig.4 TheeffectofZEB1‐AS1knockdownandcombineddoxorubicin on the apoptosis of Raji and Ramos cells
2.5 LncRNA⁃miRNA⁃mRNA调控网络的筛选与构建
通过DIANA数据库预测出4个与lncRNA高度结合的miRNA,即miR⁃33⁃5p、miR⁃222⁃3p、miR⁃499a⁃5p、miR⁃4742⁃3p,结合位点见图5A。利用R语言对芯片表达量进行预处理,并按照lncRNA ZEB1⁃AS1表达量的中位数分为高表达组和低表达组后进行mRNA共表达分析,结果共筛选出1 208个上调mRNA(与lncRNA ZEB1⁃AS1呈现共表达关系),以前50个差异基因绘制的热图见图5B。通过R语言limma包和Targetscan数据库,预测既与ZEB1⁃AS1呈共表达关系又与miRNA结合的差异表达mRNA,结果显示可能存在261个靶基因受以上4个miRNAs调控,其中miR⁃33⁃5p、miR⁃221⁃3p、miR⁃499a⁃5p 和 miR⁃4742⁃3p 的靶基因分别有38个、27个、16个、180个。结合以上lncRNA⁃miRNA⁃mRNA的调控机制,最终建立ZEB1⁃AS1在DLBCL中的lncRNA⁃miRNA⁃mRNA调控网络,包括284个节点和291条边。其中,284个节点代表1个ZEB1⁃AS1、4个miRNAs和232个mRNAs,291条边表示它们之间存在291种相互作用关系,见图5C;ZEB1⁃AS1位于该网络中心,调节与之结合的miR⁃33⁃5p、miR⁃221⁃3p和miR⁃499a⁃5p、miR⁃4742⁃3p,进而调控下游232个靶基因。
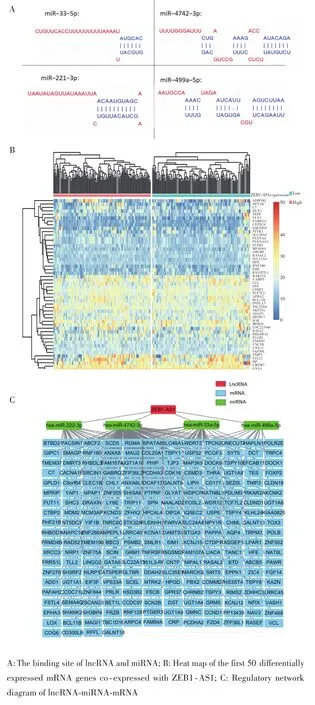
图5 LncRNA‐miRNA‐mRNA调控网络的筛选与构建Fig.5 Screening and construction of lncRNA‐miRNA‐mRNA regu‐latory network
2.6 下游基因功能及信号通路分析
GO分析共发现18个细胞成分、20个生物过程、3个分子功能相关富集,主要参与中心体、线粒体基质、蛋白酶体蛋白质分解代谢、D N A代谢调控、蛋白酶体介导的泛素依赖性蛋白分解代谢、泛素样蛋白转移酶活性等功能;KEGG分析发现6条相关通路,包括泛素介导的蛋白水解、细胞周期、内质网中蛋白加工过程、丙酸代谢等通路,见图6。在PPI网络预测中发现,R C HY1是拥有最高连接度的核心基因,与多种蛋白质存在相互作用。
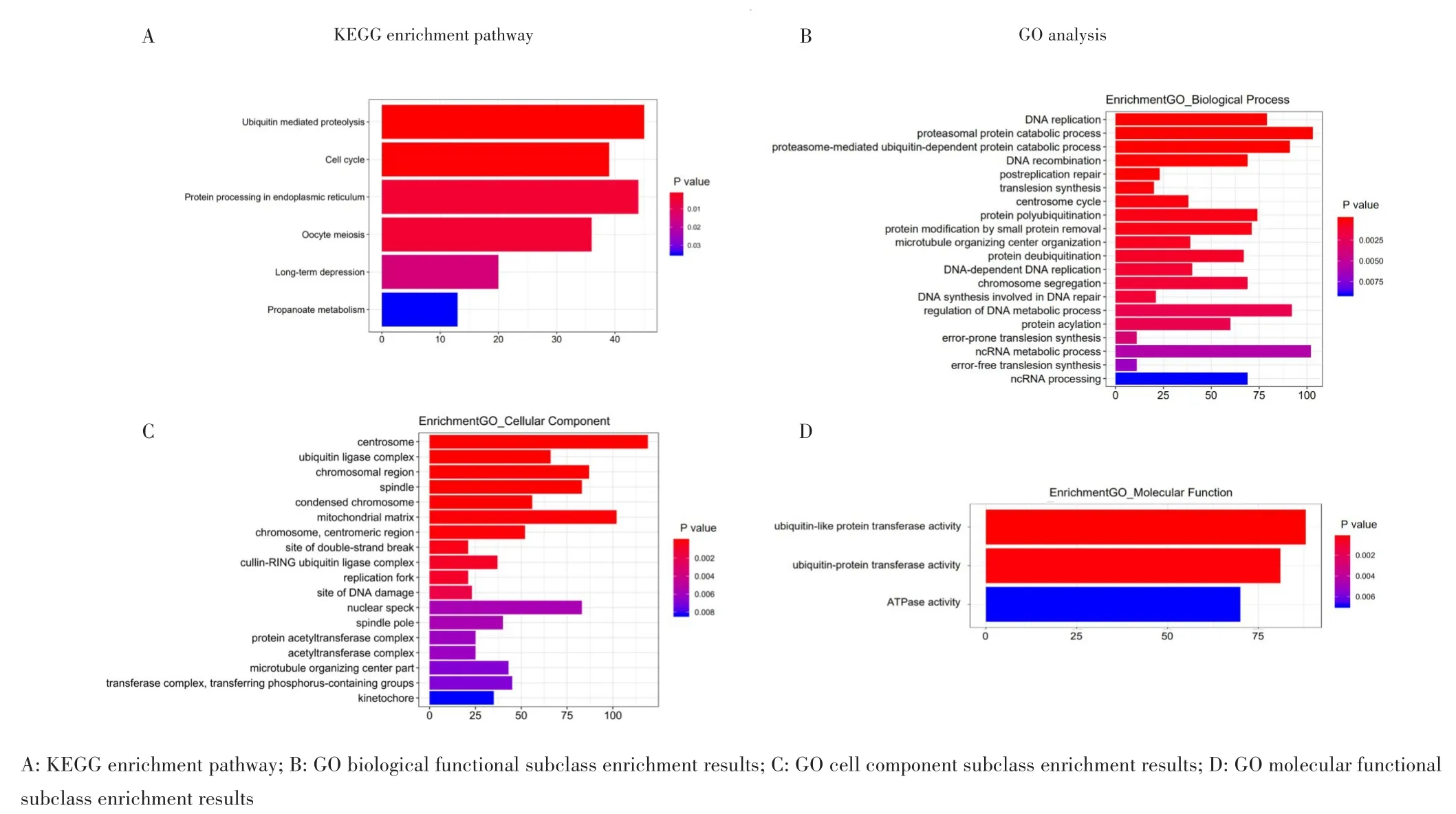
图6 LncRNA靶基因KEGG、GO分析Fig.6 KEGG and GO analysis of lncRNA target genes
3 讨论
近年来,越来越多的研究发现lncRNA可能是肿瘤发生和发展过程中的生物标志物[10],如在直肠癌中发现lncRNAs有良好的诊断和预后价值[11]。更重要的是,lncRNA还能通过参与调控肿瘤生物过程,结合下游靶基因并且调节其功能,进而影响增殖、凋亡以及分化等生物过程,在肿瘤发生发展、预后和治疗中发挥重要的生物调控作用。例如,在肝癌中发现lncRNA WT1⁃AS通过上调WT1蛋白含量抑制细胞凋亡[12]。但是目前大多数lncRNAs功能尚不明确,其中lncRNA ZEB1⁃AS1与淋巴瘤也未见相关报道。本研究通过GEO数据集和qPCR验证lncRNA ZEB1⁃AS1在B⁃NHL细胞中高表达,且高表达预示不良生存结局。该基因的转录本从具有锌指结构E⁃box⁃结合同源框1(zinc finger E⁃box binding homeobox 1,ZEB1)的共享双向启动子转录,其转录物可结合赖氨酸甲基转移酶2A,并促进组蛋白修饰,而该修饰被认为可促进ZEB1表达。已有研究发现ZEB1⁃AS1可诱导肝细胞癌上皮⁃间充质转化和远处转移[13];上调ZEB1⁃AS1表达可促进骨肉瘤细胞增殖和迁移[14]。本研究在体外实验通过基因敲降联合Dox处理进一步验证ZEB1⁃AS1功能。细胞增殖实验结果也发现,敲低ZEB1⁃AS1能抑制B⁃NHL细胞增殖,且相较单独Dox及单独敲低ZEB1⁃AS1,Dox同时处理ZEB1⁃AS1敲降细胞后的增殖抑制效果更明显,提示敲低ZEB1⁃AS1能减弱B⁃NHL细胞系增殖能力,且可能对Dox的化疗杀伤起协同作用。流式细胞术实验也证实敲低ZEB1⁃AS1能促进B⁃NHL细胞凋亡,敲低ZEB1⁃AS1可以协同Dox减少淋巴瘤对Dox的抵抗和耐受。机制可能与lncRNA能通过多种调节机制在细胞转录、转录后水平以及染色体修饰等方面发挥调节作用有关[15]。
ceRNAs是一种lncRNA调控模式,在基因表达调节中具有重要作用,参与肿瘤发生及进展[16⁃18]。在ceRNAs网络中lncRNA以miRNA作为桥梁间接调控蛋白编码基因表达[5]。已有研究报道lncRNA ZEB1⁃AS1为节点的调控网络,如靶向miR⁃365a⁃3p抑制肝癌细胞增殖[19];通过竞争性结合 miR⁃141⁃3p促进肺纤维化[17]。本研究结果也显示lncRNA ZEB1⁃AS1是B⁃NHL ceRNA网络的上游信号分子,且发挥类似海绵作用,其中miR⁃33⁃5p、miR⁃221⁃3p、miR⁃499a⁃5p和miR⁃4742⁃3p可能与下游261个靶基因形成调控网络的miRNA,但是lncRNA ZEB1⁃AS1与以上4个miRNA的调控关系有待深入研究。
本研究进一步进行信号通路分析及分子功能预测,结果发现lncRNA的靶基因与中心体、线粒体基质、蛋白酶体蛋白质分解代谢、DNA代谢调控、泛素样蛋白转移酶活性等通路显著相关。将这些在信号通路中富集的分子导入PPI网络,发现RCHY1具有最高的连接度且与多种蛋白质存在相互作用,为寻找下游调控靶点提供基础。
综上所述,ZEB1⁃AS1敲低可抑制B⁃NHL细胞增殖和促进细胞凋亡,ZEB1⁃AS1敲低协同Dox作用后的杀伤作用更明显,lncRNA ZEB1⁃AS1可能作为DLBCL潜在的生物标志物,以ZEB1⁃AS1为节点构建的ceRNA调控网络,可能是DLBCL重要的调控机制和诊疗靶点。
