采用重叠PCR构建高灵敏度酵母细胞传感器评估遗传毒性化合物
2021-05-08夏星雅魏嘉利
何 颖,夏星雅,魏嘉利,郑 枫*
(1中国药科大学药物分析学教研室;2教育部药品安全与预警重点实验室,南京210009)
遗传毒性化合物由于其潜在的致突变性与致癌作用,引起了制药行业和监管机构的高度关注[1-2]。遗传毒性由化合物与DNA或其他控制细胞凋亡的靶标相互作用引起,包括了DNA加合物的诱导、链断裂、点突变以及染色体的结构和数值变化[3-5]。通常使用体内啮齿动物致癌性试验准确评估化合物的致癌性,对于没有致癌性数据或支持性致癌数据的化合物,则采用基因毒性试验来预测致癌潜力[6-8]。自20世纪70年代以来,已经开发了几种基于细菌的生物测定法来评估化合物潜在的遗传毒性以作为动物试验的替代[9]。Ames试验是致突变性短期检测最常用的方法之一,其原理基于监测鼠沙门氏菌突变株中诱变剂引起的逆突变[8]。Ames试验具有易操作、成本低的优点,但由于细菌检测在药物代谢、膜转运和DNA修复系统方面与真核生物存在差异,试验会产生大量假阳性与假阴性结果[10]。目前已经开发几种基于哺乳动物细胞的微核试验和彗星实验来代替动物测试,但均存在评估成本过高和评估时间过长的不足[11]。因此,亟需发展一种成本更低、速度更快、信息可靠的遗传毒性评估方法。
芽殖酵母酿酒酵母(Saccharomyces cerevisiae)是一种代表性的单细胞真核生物,在遗传毒性测试应用方面具有多种优势。酵母细胞生长迅速、易于培养、遗传背景简单,使其成为优良的生物传感工具[12]。同时,作为真核生物,酵母与人类在许多代谢通路和蛋白表达调节等方面高度保守[13]。当遗传毒性化合物引起酵母细胞DNA损伤时,细胞通过DNA损伤检查点通路激活一系列蛋白激酶来进行修复,相关基因的转录水平被上调,因此可以通过测量转录水平来评估DNA的损伤水平[14-15]。核糖核苷酸还原酶(ribonucleotide reduc⁃tase,RNR)是催化核糖核酸转变为脱氧核糖核苷酸过程的限速酶,在DNA的复制与修复过程中发挥重要作用[16]。酵母细胞中存在4种亚单位的RNR(RNR1、RNR2、RNR3、RNR4),其中RNR2在正常状态下表达水平很低,当受到DNA损伤诱导后,蛋白表达增加十几倍[17]。因此,将RNR2启动子(pRNR2)作为感应元件与报告元件酵母增强绿色荧光蛋白(yeast enhanced green fluorescent pro⁃tein,yEGFP)基因相融合,构建pRNR2调控的酵母重组载体(pRNR2-yEGFP),并将重组载体转入酵母细胞中构建细胞传感器。当遗传毒性化合物作用于该重组酵母细胞传感器时,能够诱导pRNR2-yEGFP元件中的荧光报告基因的表达,通过测量荧光强度实现对化合物遗传毒性的定量评估。
通过采用细胞壁合成与药物转运突变体改善酵母细胞渗透性,已经实现酵母细胞传感器的检测灵敏度与特异性的提高,敲除细胞壁合成基因erg6、cwp1和cwp2与膜转运蛋白基因pdr5、snq2和yor1是提高酵母细胞传感器检测遗传毒性灵敏度的有效方法[18-19]。酵母细胞具有较高的同源重组频率和较短的同源片段长度需求,因此采用40 bp的同源臂的一步PCR产物转化能够进行目标基因的敲除[20]。为了提高酵母细胞基因敲除的效率,需要设计更长的同源臂,而一步PCR产物转化法难以满足,通常需要采取酶切、连接的方法构建基因敲除组件[19]。本研究采用重叠PCR(overlap PCR)构建pdr5与snq2基因敲除组件,设计的同源臂长度为600~700 bp,相较于传统方法具有成本更低、操作简便的优点。构建了pRNR2调控的基因突变型酵母细胞传感器对遗传毒性化合物进行定量评估,并比较了不同基因突变对酵母细胞传感器检测准确度与灵敏度的影响。
1材料
1.1 质粒与菌株
酵母重组质粒pRNR2-yEGFP由扬州大学医学院预防医学系李湘鸣教授赠送;大肠埃希菌DH5α感受态细胞(上海生工生物工程有限公司);酿酒酵母BY4741、pESC-Leu质粒、pESC-His质粒(上海柯雷生物科技有限公司)。
1.2试剂
SD/-Ura、SD/-Leu、SD/-His、SD/-His-Leu、SD/-Ura-Leu、SD/-Ura-His、SD/-Ura-His-Leu培养基(上海艾礼生物科技有限公司);PrimeSTAR Max DNA聚合酶(日本TaKaRa生物股份有限公司);PCR产物纯化试剂盒、无毒核酸染料、TE缓冲液、TAE缓冲液(上海生工生物工程有限公司);质粒DNA小提试剂盒、胶回收/DNA纯化试剂盒(南京诺唯赞生物科技有限公司);DNA上样缓冲液(南京翼飞雪生物科技有限公司);甲磺酸甲酯(methyl meth⁃anesulfonate,MMS,98%,批号:EJ120007)、甲磺酸乙 酯(ethyl methanesulfonate,EMS,98%,批 号:DK190025)(上海阿拉丁生化科技股份有限公司);顺铂(65%)、4-硝基喹啉-N-氧化物(4-nitroquino⁃line-N-oxide,4NOQ,98%)、5-氟尿嘧啶(5-fluoroura⁃cil,5-FU,99%)、羟基脲(99%)、水杨酸(99.5%)、葡萄糖(99%)(上海麦克林生化科技有限公司);其他试剂均为市售分析纯。
1.3仪器
专业型梯度PCR仪(德国耶拿公司);Spectra⁃Max M2e多功能酶标仪(美国分子仪器公司);ECLIPSE Ti2-U型倒置荧光显微镜(日本尼康公司);3500全自动数码凝胶图像分析系统(上海天能科技有限公司)。
2方 法
2.1 pdr5Δ∷LEU2与snq2Δ∷HIS3基因敲除组件的构建
按常规玻璃珠法从酿酒酵母BY4741中提取基因组DNA后,采用overlap PCR构建基因敲除组件,原理如图1,研究中使用的引物见表1。首先PCR扩增pdr5上游同源臂片段,设计引物q-1/q-2,PCR产物经DNA纯化试剂盒回收,命名为pdr5-1。同法扩增pdr5下游同源臂片段,设计引物q-3/q-4,得到纯化产物pdr5-2。以pESC-Leu质粒为模板,扩增LEU2基因片段,设计引物q-5/q-6,得到纯化产物LEU2。
采用overlap PCR方法构建pdr5Δ∷LEU2基因敲除组件。分别设计引物q-1/q-6与q-4/q-5,采用overlap PCR连接pdr5-1与LEU2片段以及pdr5-2与LEU2片段,分别得到纯化产物pdr5-1-LEU2与pdr5-2-LEU2。设计引物q-7/q-8,采用overlap PCR连接pdr5-1-LEU2与pdr5-2-LEU2片段,得到纯化产物pdr5Δ∷LEU2。
采用类似方法构建snq2Δ∷HIS3基因敲除组件。首先设计引物q-9/q-10、q-11/q-12与q-13/q-14,分别扩增得到snq2上下游同源臂snq2-1、snq2-2以及HIS3片段;设计引物q-9/q-14与q-12/q-13采用overlap PCR扩增分别得到snq2-1-HIS3与snq2-2-HIS3片段;设计引物q-15/q-16,采用overlap PCR连接snq2-1-HIS3与snq2-2-HIS3片段,纯化后得到snq2Δ∷HIS3。

Table1 PCRprimersused in thestudy
2.2 基因突变酵母细胞的构建
采用醋酸锂转化法将pdr5Δ∷LEU2与snq2Δ∷HIS3基因敲除组件分别转入野生型酵母细胞BY4741,经SD/-Leu与SD/-His平板筛选得到pdr5单基因突变与snq2单基因突变酵母细胞;再将snq2Δ∷HIS3基因敲除组件转入snq2单基因突变酵母细胞,经SD/-His-Leu平板筛选得到pdr5、snq2双基因突变酵母细胞。采用玻璃珠法提取pdr5单基因突变、snq2单基因突变与pdr5、snq2双基因突变酵母细胞的基因组DNA,分别设计引物q-7/q-8、q-15/q-16与q-7/q-8和q-15/q-16扩增突变片段后进行测序验证。
2.3 pRNR2调控的重组酵母细胞传感器的建立
采用醋酸锂转化法将pRNR2-yEGFP转化于感受态酵母细胞BY4741,经SD/-Ura、SD/-Ura-Leu、SD/-Ura-His、SD/-Ura-His-Leu平板筛选得到pRNR2调控的野生型、pdr5单基因突变、snq2单基因突变与pdr5、snq2双基因突变酵母细胞传感器。提取酵母细胞传感器质粒DNA,设计引物q-17/q-18扩增质粒片段进行验证。
2.4 最大非细胞毒性浓度的确定
使用2%DMSO水溶液作为溶剂配制遗传毒性化合物与阴性对照储备液,并逐级稀释至以下质量浓度:MMS(10,30,60,80,100,180,250,300,400µg/mL);EMS(50,100,200,400,800,1 000,1 300,1 500,2 000µg/mL);顺铂(10,20,30,40,60,90,120,150,200µg/mL);4NOQ(0.01,0.02,0.05,0.1,0.2,0.5,1,2,5µg/mL);5-FU(0.01,0.05,0.1,0.2,0.4,0.6,0.8,1,2µg/mL);羟基脲(100,200,400,500,800,1 200,1 500,2 000,2 500µg/mL);水杨酸(0.1,0.5,1,2,5,10,15,30,50µg/mL);葡萄糖(5,10,20,40,60,80,100,150,200µg/mL)。过夜培养4种酵母细胞单克隆,用新鲜的酵母培养液稀释至A600为0.1。以底部透明的黑色微孔板为反应载体,每孔中加入稀释后酵母培养液190µL与测试化合物溶液或溶剂对照10µL,每种质量浓度设置3个平行对照。微孔板置于30℃,220 r/min环境下培养24 h后,使用酶标仪测量A600。最终测定时每孔中DMSO的浓度低于0.1%,对酵母细胞生长没有明显影响。
2.5 最佳暴露时间的确定
过夜培养4种酵母细胞单克隆,用新鲜的酵母培养液稀释至A600为0.1。以底部透明的黑色微孔板为反应载体,每孔中加入稀释后酵母培养液190µL与MMS溶液10µL,质量浓度为10,30,60,80,100,150,180,200,250µg/mL,每种浓度设置3个平行对照。微孔板置于30℃,220 r/min环境下培养,使用酶标仪每间隔4小时测量A600与EGFP强度(激发光波长485 nm,发射光波长535 nm),持续时间20 h。
2.6 DNA损伤试剂暴露实验
根据实验“2.4”与“2.5”项下确定测试化合物溶液的最大非细胞毒性浓度与细胞传感器的最佳暴露时间,选用适宜浓度测试化合物溶液孵育4种酵母细胞传感器适当时间:MMS(10,30,60,100,150,200,250µg/mL);EMS(50,100,300,600,1 000,1 300,1 500µg/mL);顺铂(10,20,30,60,90,120,150µg/mL);4NOQ(0.01,0.05,0.1,0.2,0.5,0.7,1µg/mL);5-FU(0.01,0.05,0.1,0.15,0.2,0.3,0.4µg/mL);羟基脲(100,200,400,800,1 000,1 200,1 500µg/mL);水杨酸(1,2,5,10,15,20,30µg/mL);葡萄糖(10,20,40,80,100,150,200µg/mL)。每种质量浓度设置3个平行对照,其余操作同“2.5”项。
2.7 数据统计和分析
在“2.4”项下中,采用Microsoft Excel 2013软件计算测试化合物各浓度下4种酵母细胞传感器A600均值,取3次独立实验的平均值和标准差。定义Ax/A0为暴露组A600与溶剂对照组A600的比值,设定Ax/A0小于90%为遗传毒性数据的排斥阈值。
在“2.5”与“2.6”项中,通过扣除空白培养基校正EGFP与A600,采用Microsoft Excel 2013软件计算4种酵母细胞传感器的EGFP与A600的均值(n=3)。定义单位细胞的平均相对荧光强度AF(aver⁃age fluorescence)=EGFP/A600,取3次独立实验的平均值及标准差。定义荧光诱导倍数FI(fold induc⁃tion)为暴露组与溶剂对照组的单位细胞的平均相对荧光强度之比,则FI=AFtreated/AFuntreated。设定遗传毒性阈值为1.5倍,即当AF增加50%,且荧光强度随剂量的增加表现出增长趋势时,判断化合物为遗传毒性阳性,反之为阴性。采用Graphpad Prism 7.00软件进行制图分析。
3结 果
3.1 pdr5Δ∷LEU2与snq2Δ∷HIS3基因敲除组件的构建
采用overlap PCR方法构建pdr5Δ∷LEU2与snq2Δ∷HIS3基因敲除组件,PCR扩增产物电泳结果见图2与图3。结果显示,pdr5Δ∷LEU2与snq2Δ∷HIS3基因敲除组件的碱基大小与预期一致,表明pdr5Δ∷LEU2与snq2Δ∷HIS3基因敲除组件构建成功。
3.2 pRNR2调控的野生型、pdr5单基因突变、snq2单基因突变与pdr5、snq2双基因突变酵母细胞传感器的构建
pdr5Δ∷LEU2与snq2Δ∷HIS3基因敲除组件转入酵母细胞后,经平板筛选得到的单克隆DNA的PCR扩增产物电泳结果见图4与图5。结果显示,扩增出的酵母细胞基因突变片段的碱基大小与预期一致。对PCR扩增产物进行测序并与GenBank序列比对,pdr5基因片段相似度为99.88%(3 289/3 293),间隙为0.12%(4/3 293);snq2基因片段相似度为99.96%(2 542/2 543),间隙为0.04%(1/2 543),表明pdr5单基因突变、snq2单基因突变与pdr5、snq2双基因突变酵母细胞构建成功。

Figure 2 Gene knockout of pdr5Δ∷LEU2M:DNA markers;Lane 1-2:pdr5-1-LEU2(2 789 bp);Lane 3-4:pdr5-2-LEU2(2 817 bp);Lane5-9:pdr5Δ∷LEU2 geneknockout(3 304 bp)
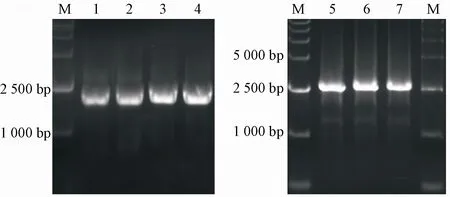
Figure 3 Gene knockout of snq2Δ∷HIS3M:DNA markers;Lane 1-2:snq2-1-HIS3(1899 bp);Lane 3-4:snq2-2-HIS3(1 948 bp);Lane5-7:snq2Δ∷HIS3 gene knockout(2540 bp)
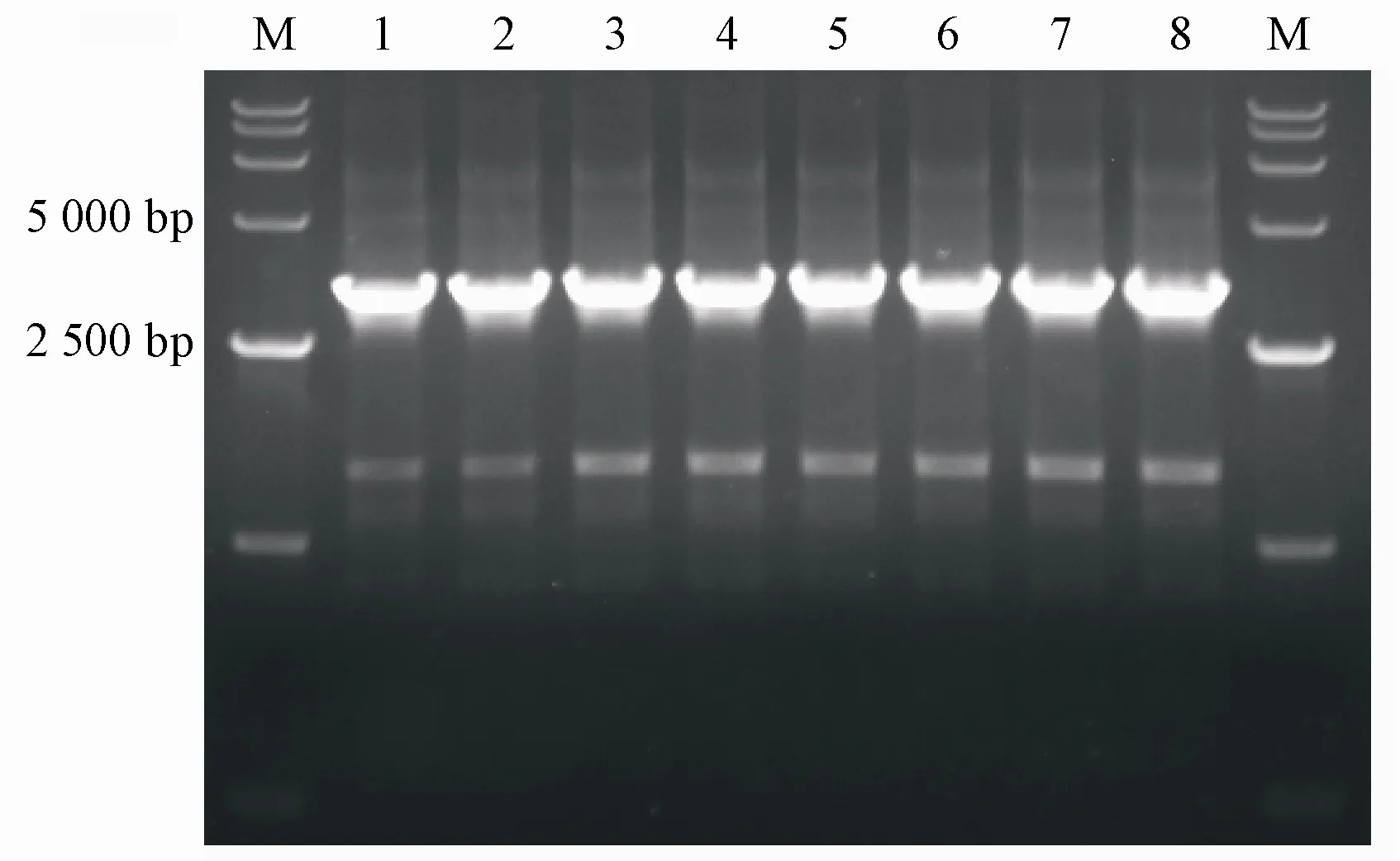
Figure 4 Mutant gene fragment of pdr5M:DNA markers;Lane 1-4:Single-gene mutation of pdr5(3 293 bp);Lane5-8:Double-genemutation of pdr5 and snq2(3 293 bp)
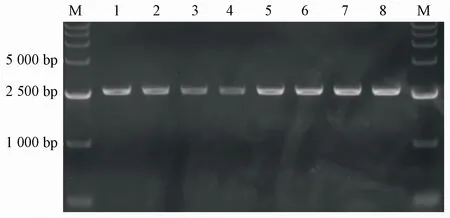
Figure 5 Mutant gene fragment of snq2M:DNA markers;Lane 1-4:Single-gene mutation of snq2(2 543 bp);Lane5-8:Double-genemutation of pdr5 and snq2(2 543 bp)
pRNR2-yEGFP重组载体转入野生型、pdr5单基因突变、snq2单基因突变与pdr5、snq2双基因突变4种酵母细胞,质粒DNA的PCR扩增产物电泳结果见图6。结果显示,扩增出的质粒DNA片段的碱基大小与预期一致,表明重组载体pRNR2-yEGFP已转入4种酵母细胞,pRNR2调控的野生型、pdr5单基因突变、snq2单基因突变与pdr5、snq2双基因突变酵母细胞传感器构建成功。
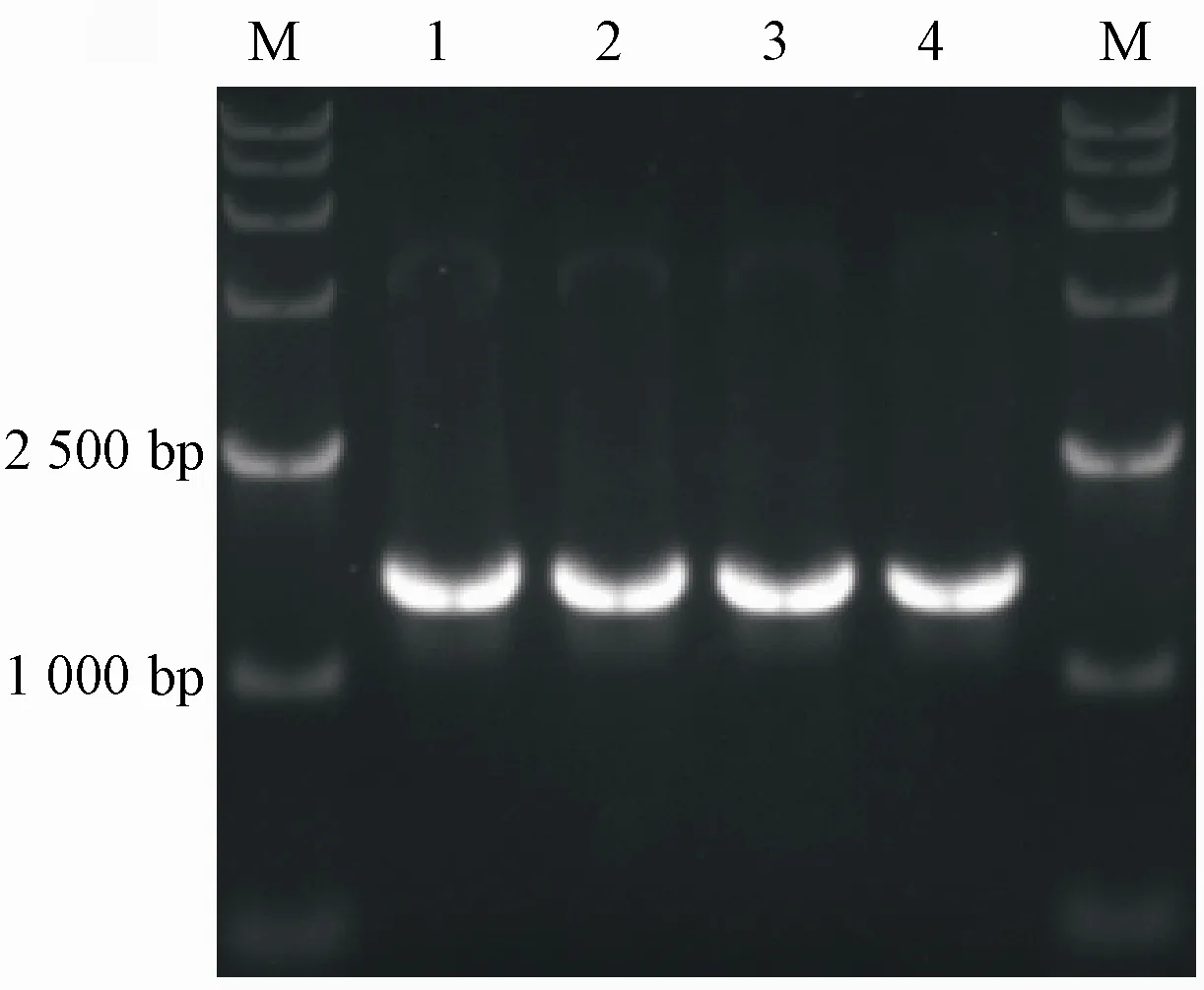
Figure6 Fragment of recombinant plasmid pRNR2-yEGFPM:DNA markers;Lane 1:Wild-type yeast cell sensor(1 493 bp);Lane 2:Yeast cell sensor of single-gene mutation of pdr5(1 493 bp);Lane 3:Yeast cell sensor of single-gene mutation of snq2(1 493 bp);Lane 4:Yeast cell sensor of double-gene mutation of pdr5 and snq2(1 493 bp)
3.3 化合物对酵母细胞传感器的最大细胞毒性浓度的确定
应用野生型、pdr5单基因突变、snq2单基因突变与pdr5、snq2双基因突变酵母细胞传感器对8种测试化合物进行24 h生长抑制测试,4种细胞传感器的Ax/A0结果见图7。结果显示,MMS、EMS、顺铂、4NOQ、5-FU、羟基脲与阴性对照水杨酸在高浓度下表现出抑制细胞生长的细胞毒性,阴性对照葡萄糖未表现出明显细胞毒性。
3.4 MMS对酵母细胞传感器最佳暴露时间的确定
经“2.4”项确定MMS最大非细胞毒性浓度后,采用MMS进行最佳暴露时间测试,野生型、pdr5单基因突变、snq2单基因突变与pdr5、snq2双基因突变4种细胞传感器结果见图8。当pRNR2调控的细胞传感器与MMS作用时,FI值随时间增加而逐渐增大,并于16 h时达到最大值,继续暴露至20 h时下降,因此选择16 h作为酵母细胞传感器的暴露时间。
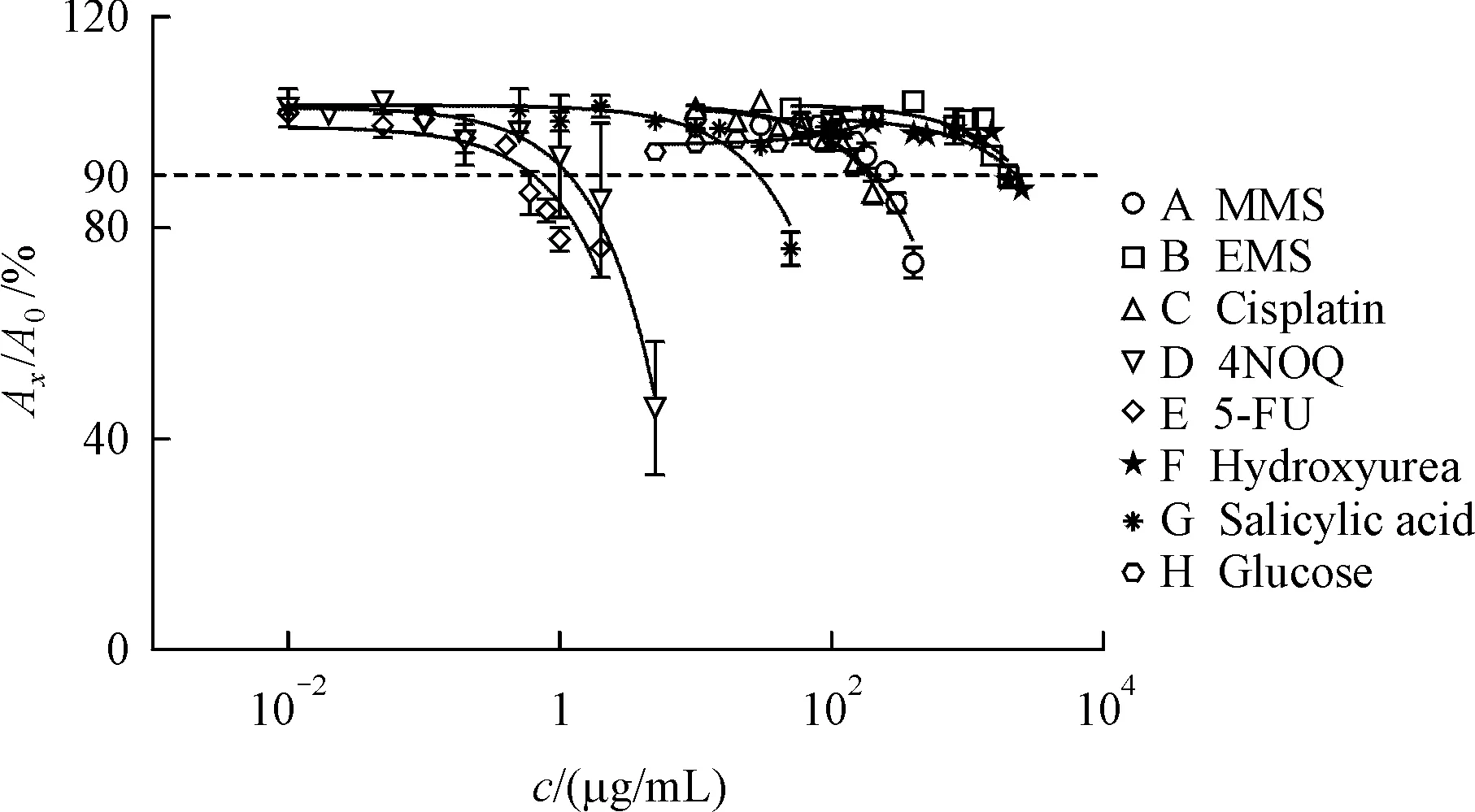
Figure 7 Cytotoxicity in yeast cells for tested compounds in 24 h(xˉ±s,n=3)A:MMS(methyl methanesulfonate);B:EMS(ethyl methanesulfonate);C:Cisplatin;D:4NOQ(4-nitroquinoline-N-oxide);E:5-FU(5-fluoroura⁃cil);F:Hydroxyurea;G:Salicylic acid;H:Glucose
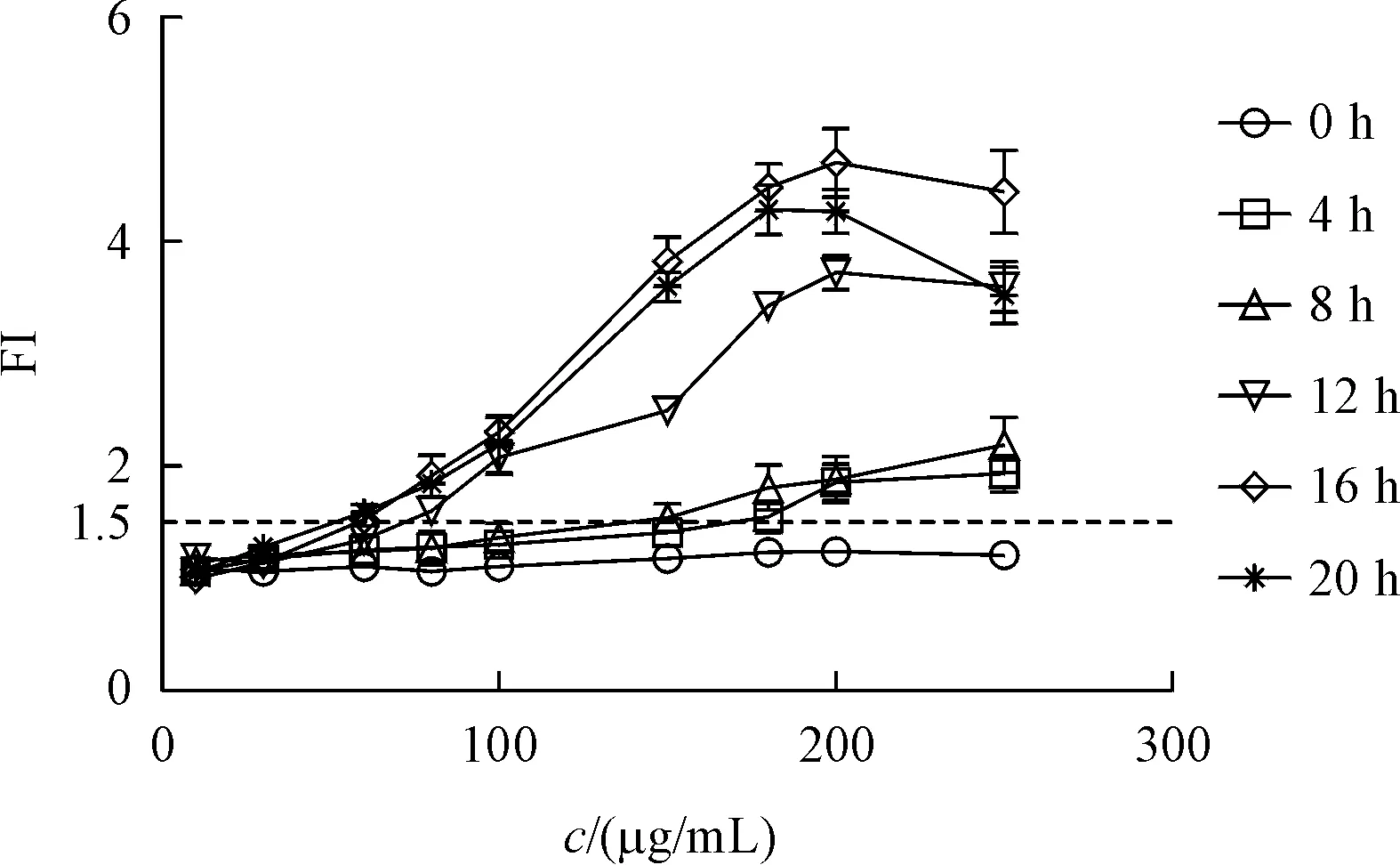
Figure 8 Yeast cell sensors exposed to different concentrations of MMSsolution for 0,4,8,12,16,20 h(xˉ±s,n=3)
当MMS质量浓度最低为50µg/mL时,16 h暴露组细胞传感器FI值超过1.5。pRNR2调控的野生型细胞传感器暴露于MMS溶液下16 h的倒置荧光显微镜图见图9。与溶剂对照组(图9-C)相比,50µg/mL MMS溶液组(图9-D)的细胞传感器表达出更亮的荧光。结果进一步表明16 h为细胞传感器适宜的暴露时间。
3.5 酵母生物传感器暴露于测试化合物的荧光蛋白表达的量效关系
野生型、pdr5单基因突变、snq2单基因突变与pdr5、snq2双基因突变4种酵母细胞传感器暴露于系列浓度测试化合物16 h后,荧光蛋白表达的剂效关系见图10。将本次研究中酵母细胞传感器测试结果与已有报道的Ames试验、体外遗传毒性试验与体内遗传毒性试验的数据进行对比,结果如表2所示。对于体内外遗传毒性数据呈阴性的两种测试化合物水杨酸与葡萄糖,4种酵母细胞传感器检测结果均表现为遗传毒性阴性;对于已有体内外遗传毒性数据的6种测试化合物,snq2单基因突变与pdr5、snq2双基因突变细胞传感器均检测出阳性结果,测试准确度100%,野生型与pdr5单基因突变细胞传感器能检测出4NOQ以外的其余五种测试化合物,测试准确度87.5%。结果表明,pRNR2调控的细胞传感器能够用于遗传毒性化合物检测,且snq2单基因突变与pdr5、snq2双基因突变细胞传感器表现出更高的准确度与特异性。
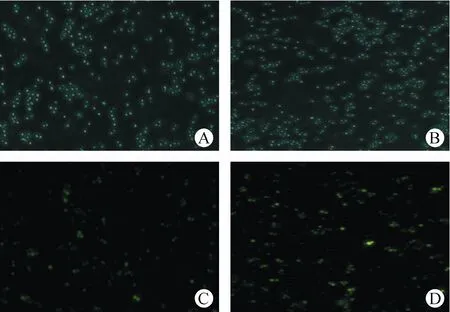
Figure 9 Wild-type cell sensor regulated by pRNR2 exposed to MMS solution for 16 hA:Bright-field image at 0µg/mL;B:Bright-field image at 50µg/mL;C:Inverted fluorescence microscope image at 0µg/mL;D:Inverted fluo⁃rescence microscope image at 50µg/mL
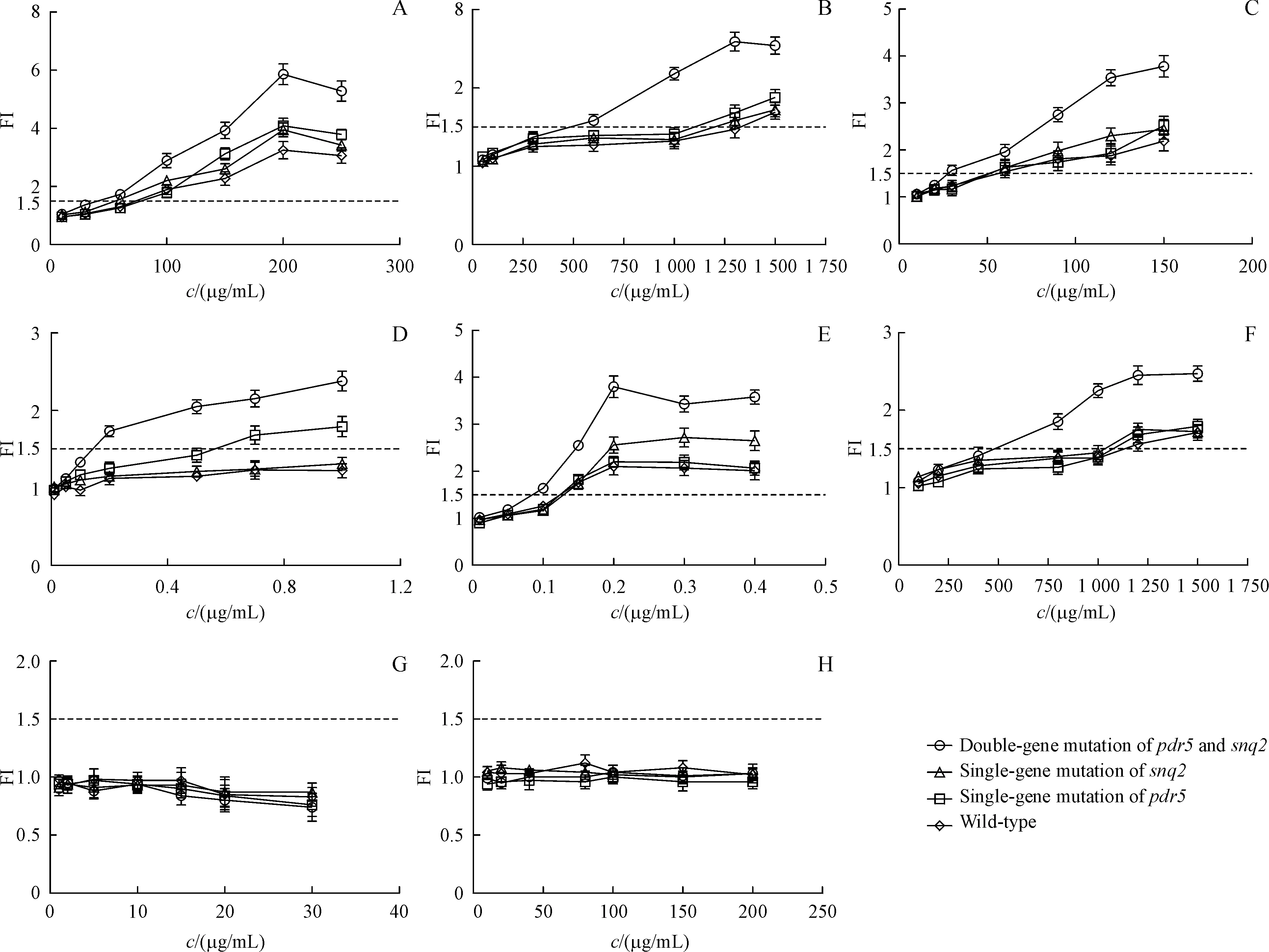
Figure10 Yeast cell sensorsexposured totested compoundsfor 16 h(xˉ±s,n=3)A:MMS(methyl methanesulfonate);B:EMS(ethyl methanesulfonate);C:Cisplatin;D:4NOQ(4-nitroquinoline-N-oxide);E:5-FU(5-fluorouracil);F:Hydroxyurea;G:Salicylic acid;H:Glucose
对于测试的6种遗传毒性阳性模型化合物,pdr5、snq2双基因突变细胞传感器表现出明显高于其他3种细胞传感器的FI。在检测灵敏度方面,双基因突变细胞传感器同样表现出明显优势。在测试6种遗传毒性数据阳性化合物时,相较于其他3种细胞传感器,双基因突变细胞传感器达到遗传毒性阈值所需浓度降低了数倍。snq2单基因突变细胞传感器相较于pdr5单基因突变细胞传感器,在检测4NOQ中表现出更高的准确度;而在检测5-FU方面,pdr5单基因突变细胞传感器表现出更高的荧光响应。野生型细胞传感器在检测灵敏度与荧光响应强度方面并无突出表现。结果表明尽管snq2与pdr5高度同源,具有共同的反应底物,但对于特异性底物表现出不同的响应强度,snq2单基因突变细胞传感器的检测准确度优于pdr5单基因突变细胞传感器,双基因突变细胞传感器在测试中表现出最佳的准确度和灵敏度。
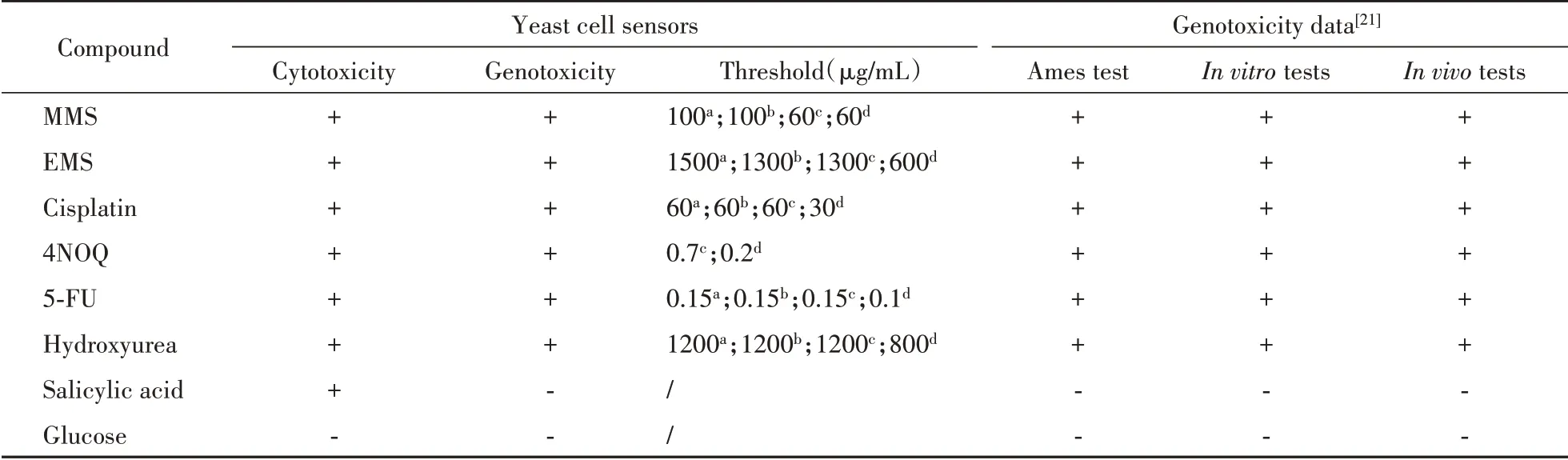
Table2 Genotoxicity of compoundstested by yeast cell sensorsand comparison with other tests
4讨论
基于DNA损伤诱导型启动子的酵母细胞传感器进行遗传毒性测定具有成本低、样品用量少、检测迅速、准确度高的优点,已被广泛用于环境样品与药物化合物的遗传毒性评估。通过敲除膜转运蛋白基因或细胞壁合成基因增加酵母细胞渗透性已成为改善细胞传感器检测灵敏度的有效方法。本研究采用overlap PCR方法分别构建pdr5与snq2基因敲除组件pdr5Δ∷LEU2与snq2Δ∷HIS3,设计的上下游同源臂长度为600~700 bp。将基因敲除组件与pRNR2-yEGFP重组质粒通过醋酸锂转化法转入酵母细胞得到野生型、pdr5单基因突变、snq2单基因突变与pdr5、snq2双基因突变酵母细胞传感器,PCR扩增产物电泳结果与基因测序结果验证了基因突变细胞传感器构建的成功。采用24 h酵母生长抑制试验测得各化合物最大非细胞毒性浓度,测试的6种遗传毒性数据阳性化合物MMS、EMS、顺铂、4NOQ、5-FU、羟基脲与阴性化合物水杨酸都在一定浓度下表现出细胞毒性,遗传毒性数据阴性化合物葡萄糖在测试浓度范围内未表现出细胞毒性。4种酵母细胞传感器暴露于系列浓度化合物16 h后,结果表明snq2单基因突变与pdr5、snq2双基因突变细胞传感器检测准确度为100%,高于野生型与pdr5单基因突变细胞传感器(87.5%)。同一浓度下,双基因突变细胞传感器的荧光响应显著高于其他3种细胞传感器,pdr5单基因突变细胞传感器在5-FU检测中表现出显著高于野生型和snq2单基因突变细胞传感器的荧光响应。结果表明尽管snq2与pdr5高度同源,蛋白功能具有代偿性,本研究发现snq2单基因突变细胞传感器的检测准确度优于pdr5单基因突变细胞传感器,双基因突变细胞传感器检测准确度与灵敏度优于单基因突变和野生型细胞传感器。
综上,本研究采用overlap PCR方法构建组件对酵母细胞进行基因敲除,相较于一步PCR产物转化法基因敲除效率更高,相较于酶切连接构建法具有成本更低、操作更简便的优势。snq2单基因突变细胞传感器的遗传毒性检测准确度优于pdr5单基因突变细胞传感器,pdr5、snq2双基因突变酵母细胞传感器具有最佳的准确度和灵敏度。因此,本实验为构建高准确度与灵敏度的酵母细胞传感器提供了思路与方法,为酵母细胞膜转运蛋白基因pdr5与snq2的进一步功能研究奠定了基础。
