HPLC法同时测定对乙酰氨基酚口服混悬液中有关物质及防腐剂
2021-05-07北京市平谷区食品药品安全监控中心101200范保瑞张悦张成志
北京市平谷区食品药品安全监控中心(101200)范保瑞 张悦 张成志
上海强生制药有限公司(200245)王正中
对乙酰氨基酚是世界卫生组织推荐的儿童最常用的解热镇痛药之一[1],常用的剂型有片剂、胶囊、口服混悬液及小容量注射液。对乙酰氨基酚口服混悬液目前国内仅有上海强生制药有限公司生产,为红色混悬液,黏稠,有甜味、果香味,为保证其口感状态适合儿童服用,处方中含有潜溶剂、甜味剂、着色剂、防腐剂、助悬剂等大量辅料。
因对乙酰氨基酚二聚体(杂质A)在生产、储存过程中极易产生,对氯乙酰苯胺(杂质B)为原料药合成时引入的杂质[2],具有潜在的基因毒性。此外,在现行对乙酰氨基酚原料药及其制剂的质量标准和文献中没有以上两种杂质的检测方法。因此,笔者参考相关文献[3-8],建立高效液相色谱法,排除辅料干扰并测定其中有关物质及防腐剂含量。建立的方法具有专属性强、准确、灵敏,可以用于对乙酰氨基酚口服混悬液的质量控制。
1 材料
1.1 仪器 LC20A型HPLC仪,包括SPD-M20A可变波长检测器(PDA)、Labsolutions色谱工作站(日本岛津公司);CPA225D电子天平(德国sartorius公司);SevenCompactpH计(瑞士Mettler-Toledo公司)。
1.2 药品与试剂 对乙酰氨基酚对照品(中国食品药品检定研究院,批号100018-201610,含量99.9%)、杂质A对照品(N-(5’-乙酰氨基-6,2’-二羟基-联苯)-3-乙酰胺)(Organix公司,VM-459-170,含量89.4%)、杂质B对照品(N-(4-氯苯基)乙酰胺)(Organix公司,D1225CS,含量99.7%)、苯甲酸对照品(Organix公司,2DH0331,含量100%)、对羟基苯甲酸丁酯对照品(Organix公司,2EA0365,含量100%)、乙腈、三氟乙酸均为色谱纯,水为纯化水。

附表1 梯度洗脱程序

附表2 回归方程和线性范围

附表3 回收率试验结果

附表4 样品有关物质和防腐剂测定结果表
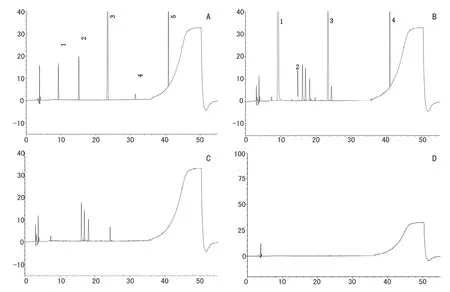
附图1 高效液相色谱图。A.对照品溶液色谱图;B.供试品溶液色谱图;C.辅料溶液色谱图;D.空白溶剂色谱图;1.对乙酰氨基酚;2.杂质A;3.苯甲酸;4.杂质B;5.对羟基苯甲酸丁酯
2 方法与结果
2.1 色谱条件 色谱柱:Hypersil GOLD C18色谱柱(4.6mm×250mm,5μm);流动相:A为0.1%三氟乙酸溶液,流动相B为0.1%三氟乙酸的乙腈溶液,梯度洗脱,流速:1.0ml/min,柱温:35℃,检测波长:245nm,进样量:20μl。梯度洗脱程序见附表1。
2.2 溶液的制备
2.2.1 供试品溶液取对乙酰氨基酚口服混悬液,剧烈振摇使混合均匀,精密称取适量(约相当于对乙酰氨基酚160mg),置100ml量瓶中,加稀释剂50ml,振摇使溶解,用稀释剂稀释至刻度,摇匀,滤过,弃去初滤液5ml,取续滤液,即得。
2.2.2 对照品溶液精密称取对乙酰氨基酚对照品约32mg、杂质A对照品约32mg和杂质B对照品约6.4mg,置同一500ml量瓶中,加入乙腈50ml振摇使溶解,并用缓冲液定容至刻度,摇匀,作为有关物质对照储备液。精密称取苯甲酸对照品约169.5mg和对羟基苯甲酸丁酯对照品约25mg,置于同一50ml量瓶中,加入乙腈5ml振摇使溶解,并用缓冲液稀释至刻度,摇匀,作为防腐剂对照储备液。精密量取有关物质对照储备液和防腐剂对照储备液各5ml,置同一200ml量瓶中,加稀释剂定容至刻度,摇匀,即得。
2.2.3 系统适用性溶液取对乙酰氨基酚口服混悬液,剧烈振摇使混合均匀,精密称取适量(约相当于对乙酰氨基酚160mg)和精密加入2.2.2项下的有关物质储备液2ml,置100ml量瓶中,加稀释剂50ml,振摇使溶解,用稀释剂稀释至刻度,摇匀,滤过,弃去初滤液5ml,取续滤液,即得。
2.2.4 灵敏度溶液精密量取对照品溶液1ml,置10ml量瓶中,用稀释剂稀释至刻度,摇匀,即得。
2.2.5 空白基质溶液取上海强生制药有限公司提供的空白辅料混合物,按2.2.1项下方法配制空白辅料溶液。
2.2.6 破坏试验储备液取对乙酰氨基酚混悬液6.5g,置100ml量瓶中,加稀释剂溶解并稀释至刻度,摇匀,即得。
2.2.7 稀释剂的配制 0.01mol/L枸橼酸缓冲液:取枸橼酸钠1.1g和枸橼酸1.3g,加水溶解并稀释至1000ml,用枸橼酸钠或枸橼酸调节pH值至(4.00±0.05)。稀释剂:0.01mol/L枸橼酸缓冲液-乙腈(90∶10)。
2.3 灵敏度及系统适用性试验 精密量取供试品溶液和对照品溶液各20μl,分别注入液相色谱仪,色谱见附图1,结果:供试品溶液中主峰与相邻杂质峰分离度均大于1.5。精密量取灵敏度溶液20μl,注入液相色谱仪,杂质B峰的信噪比不低于10,理论板数不低于5000。表明系统适用性试验结果符合要求,本方法灵敏度较高,可用于杂质定量测定。
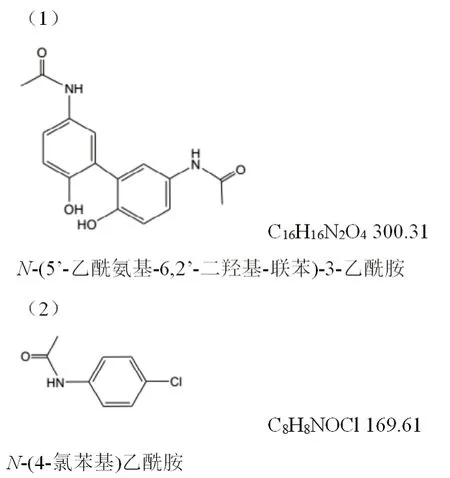
附图3 (1)杂质A(对乙酰氨基酚二聚体);(2)杂质B(对氯乙酰苯胺)
2.4 专属性试验
2.4.1 辅料干扰试验取2.2.4项下的溶液,采用上述色谱条件进样分析,结果空白辅料在本方法所考察的各物质峰处无色谱峰,辅料中泡泡糖香精、阿洛拉红Ac、品红酸D及高果糖浆的色谱峰较大,应予以扣除。故本方法采用Thermo Hypersil GOLD Aq(4.6mm×250mm,5μm)色谱柱,扣除4.5分钟前的溶剂峰以及相对保留时间约为0.8、1.7、1.8、2.0、2.6、2.7的辅料峰。
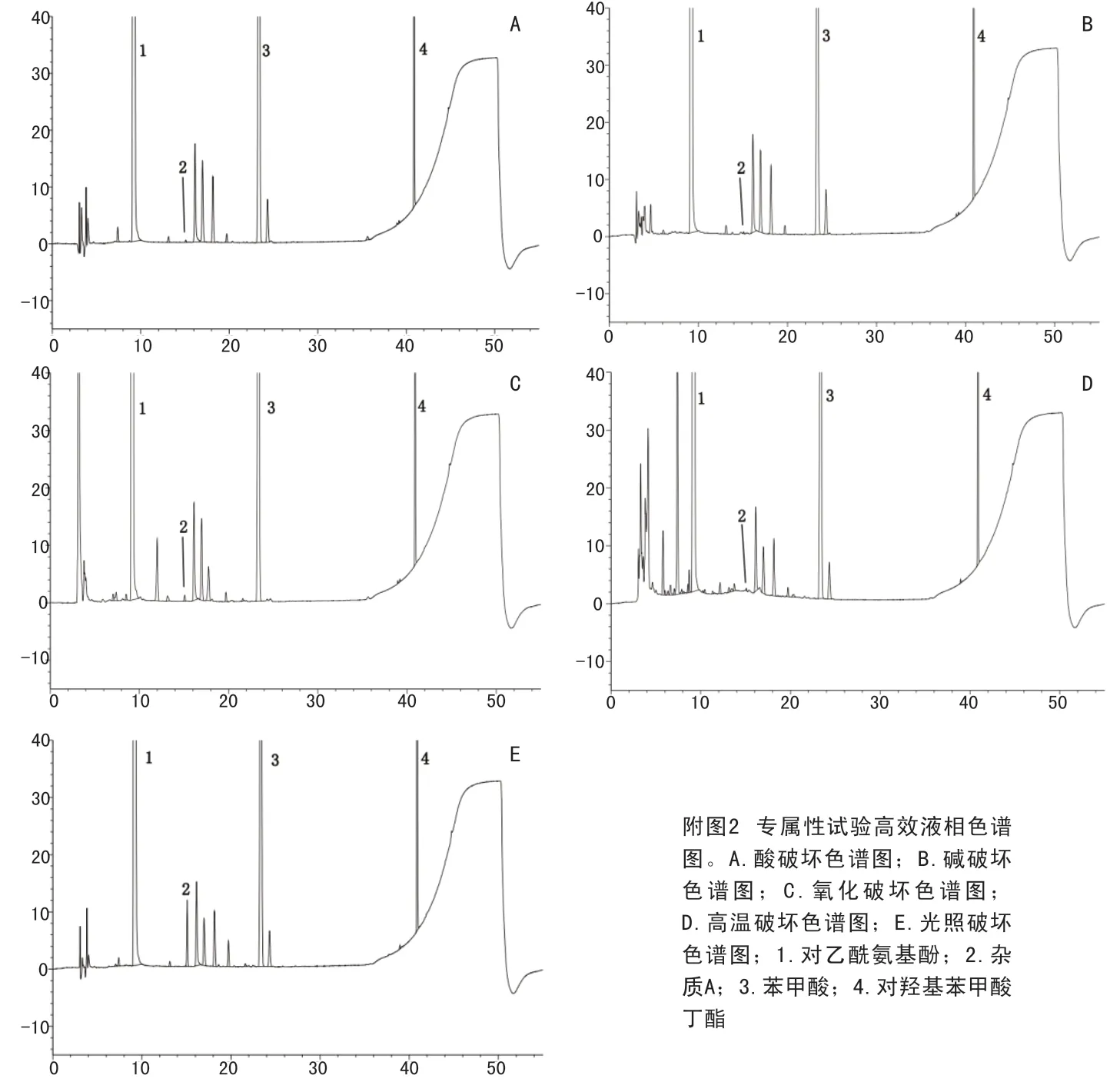
2.4.2 破坏试验 ①未破坏:取样品6.4756g,置100ml量瓶中,加稀释剂溶解并稀释至刻度,摇匀,即得。②酸破坏:取样品6.4219g,置100ml量瓶中,加1mol/L盐酸溶液6ml,室温放置5小时,加1mol/L氢氧化钠溶液6ml,加稀释剂稀释至刻度,摇匀,即得。③碱破坏:取样品6.4682g,置100ml量瓶中,加1mol/L氢氧化钠溶液6ml,室温放置5小时,加1mol/L盐酸溶液6ml,加稀释剂稀释至刻度,摇匀,即得。④氧化破坏:取样品6.4123g,置100ml量瓶中,加30%过氧化氢溶液6ml,室温放置8小时,加稀释剂稀释至刻度,摇匀,即得。⑤高温破坏:取样品6.4550g,置100ml量瓶中,置105℃放置9小时,加稀释剂溶解并稀释至刻度,摇匀,即得。⑥光照破坏:取样品6.4470g,置100ml量瓶中,置254nm紫外灯下照射9小时,加稀释剂溶解并稀释至刻度,摇匀,即得。以上各溶液按2.1项下的色谱条件进样测定。破坏性试验色谱见附图2。由附图2可见,各种因素破坏情况下,各峰分离度均大于1.5,杂质A峰纯度均大于0.99999,表明在各种破坏条件下,已知成分峰与相邻杂质分离度符合要求,方法专属性较好。
2.5 线性关系考察 精密称取对乙酰氨基酚对照品32.14mg,杂质A对照品36.18mg,杂质B对照品6.70mg,置同一500ml量瓶中,加乙腈50ml使溶解,用稀释剂稀释至刻度,混匀,作为有关物质储备液。
精密称取苯甲酸对照品170.52mg,对羟基苯甲酸丁酯对照品25.27mg,置同一50ml量瓶中,加乙腈5ml使溶解,用稀释剂稀释至刻度,摇匀,作为线性试验防腐剂储备液。
用上述两种储备液稀释制成一系列不同浓度的对照品溶液,依法测定。以检测质量浓度(x,μg/ml)为横坐标,峰面积(y)为纵坐标,进行线性回归,线性范围和回归方程见附表2。
2.6 检测限和定量限试验 将2.2项下的对照品溶液取适量流动相稀释后按2.1项下的色谱条件进样,记录色谱图,以信噪比S/N=3∶1测得杂质A、杂质B,以及防腐剂苯甲酸、对羟基苯甲酸丁酯的检测限分别为0.065ng、0.14ng、0.34ng、0.10ng;以信噪比S/N=10∶1测得各成分定量限分别为0.32ng、0.67ng、1.71ng、0.26ng。
2.7 回收率试验 精密称取对乙酰氨基酚对照品32.14mg,杂质A对照品36.18mg,杂质B对照品6.70mg,置同一500ml量瓶中,加乙腈50ml使溶解,用稀释剂稀释至刻度,混匀,作为有关物质储备液。
精密称取苯甲酸对照品170.52mg,对羟基苯甲酸丁酯对照品25.27mg,置同一50ml量瓶中,加乙腈5ml使溶解,用稀释剂稀释至刻度,摇匀,作为防腐剂储备液。
精密称取样品约6.4g,置100ml量瓶中,精密加入适量上述两种储备液,按供试品溶液配制方法进行溶解并稀释至刻度,摇匀,配制回收率试验溶液,每个浓度配制3份,共9份溶液,分别测定回收率,测定结果见附表3。
2.8 耐用性试验 笔者采用在同一台色谱仪上分别只微小改变柱温、流速、流动相中三氟乙酸浓度、梯度洗脱程序和不同色谱柱等唯一条件的方式,取2.2项下的系统适用性溶液进行试验。其中,色谱柱分别选用了Waters A tlantis T3(4.6mm×150mm,3μm);Waters XSelect HSS T3(4.6mm×250mm,5μm)和Thermo Hypersil GOLD aQ(4.6mm×250mm,5μm)在不同色谱仪上进行检测。结果:选定的色谱条件进行微调及选用不同色谱柱后,所检测到的杂质数目、出峰顺序、分离效果和杂质及防腐剂峰面积均无明显变化。
2.9 重复性试验 按照2.2.1项下方法配制6份供试品溶液(样品批号160831165),按2.1项下的方法测定,记录色谱图。结果:六份供试品测定结果一致,杂质A含量RSD=0.3%、杂质B未检出,杂质总量RSD=0.6%、苯甲酸含量RSD=0.1%、对羟基苯甲酸丁酯含量RSD=0.1%。
2.10 稳定性试验 按照2.2.1项下方法配制供试品溶液,分别在0、1、2、4、6、8、10、20h,按2.1项下的色谱条件测定,记录色谱图,以峰面积计算杂质,结果:A含量RSD=1.9%、杂质B未检出、杂质总量RSD=1.6%、苯甲酸含量RSD=0.3%、对羟基苯甲酸丁酯RSD=0.4%,表明供试品溶液在20h内稳定。
2.11 样品测定 按照2.2项下方法分别配制3份不同批号的供试品溶液,按2.1项下的方法测定,结果见附表4。
3 讨论
3.1 杂质限度的确定 本方法所测杂质为现行标准未考察的新杂质,规定限度为杂质A不得过0.1%;杂质B(对氯乙酰苯胺)为原料药合成时引入的杂质,因其具有潜在的基因毒性,故限值定为不得过0.005%。杂质分子式见附图3。
3.2 溶解方法的探索 为保证取样均一性,笔者尝试取样前要求超声和剧烈振摇两种预处理方法,考虑到超声可能导致有关物质测定结果变化,故采用剧烈振摇混合方式使样品均匀。取本品,剧烈振摇约20秒使分散均匀,称取本品约6.4g(约5ml),置100ml量瓶中,加稀释剂约50ml,振摇1小时。取出后用稀释剂稀释至刻度,摇匀,建议采用0.45μm的微孔滤膜过滤,但由于液体较黏稠,为保证过滤效果,采用8000转/分钟离心10分钟后再过滤的方式,弃去初滤液5ml,取续滤液作为供试品溶液。
3.3 检测波长的选择 制备各组分的定位溶液,采用PDA检测器,进行光谱扫描。结果表明:样品主成分对乙酰氨基酚最大吸收在243nm,已知杂质含量最低的杂质B最大吸收为246nm。将检测波长确定为主成分最大吸收和杂质B最大吸收之间且较靠近杂质B最大吸收的245nm,在此波长其他组分及破坏试验产生的杂质也有较强吸收,选定的检测波长可行。
3.4 色谱柱的选择 本方法流动相中水的比例较高,宜采用耐水性强的色谱柱,笔者考察了2.8项下的三种色谱柱进行检测。
综上所述,本方法灵敏、准确、专属性强、重复性好,可用于对乙酰氨基酚口服混悬液的质量控制。
