石墨烯量子点提高DNAzyme在细胞水平上的稳定性
2021-04-29黄奇萍张井岩
黄奇萍,刘 慧,张井岩,郑 静
(1.华东理工大学生物反应器工程国家重点实验室;2.华东理工大学药学院,上海200237)
自1994 年首次发现Pb2+依赖性切割RNA 的单链DNA 片段(又称脱氧核糖核酸酶或者DNAzyme)至今,科学家相继发现了更多具有不同催化功能的DNA⁃zyme[1⁃2]。目前发现DNAzyme 的催化功能包括:断裂和连接RNA、磷酸化DNA、氧化DNA 等。其中,催化RNA 断裂的DNAzyme 因为其潜在的应用价值而备受关注,10⁃23 DNAzyme 是其中研究最为广泛的断裂RNA 的DNAzyme 之一[3⁃4]。10⁃23 DNAzyme 是体外循环中第10轮循环的第23个克隆,由一个催化核心区域和两侧的底物结合区域组成,结合区域由DNAzyme 左右两端7~9 个和底物互补配对的脱氧核苷酸组成,催化核心区域由和底物未完全互补配对的15 个脱氧核苷酸组成。只要改变10⁃23 DNAzyme 结合区域中的碱基序列就可作用于不同的RNA 底物[5⁃6]。Cairns 等[7]根据10⁃23 DNAzyme 设计了80 多个靶向HPV⁃16 的E6、E7 mRNA 的DNAzyme 以及60 多个针 对大鼠c⁃myc 基因的DNAzyme,其中8 种DNAzyme 对HPV⁃16 致癌基因有高效的切割效率。Sun等[8]研究发现,在适宜的体外条件下,10⁃23 DNAzyme 对c⁃myc mRNA 的AU位 点 切 割 效 率 高 达20%~50%。Zhang 等[9]将靶向VEGRF⁃2的10⁃23 DNAzyme 注入到移植了肿瘤的无胸腺裸鼠体内后,裸鼠肿瘤生长得到明显的抑制。与对照组(注射生理盐水)相比,注射4 次DNAzyme 后的裸鼠肿瘤体积缩小近75%,且肿瘤外区域细胞死亡显著增加。由此可见,10⁃23 DNAzyme 作为一个潜在RNA切割工具具有很好的应用前景,唯一不足的是它在细胞水平的稳定性较差。
石墨烯量子点(GQDs)具有单层碳原子,尺寸较小,边缘具有羧基官能团,而且具有低细胞毒性[10]、良好的水溶性、化学惰性[11]等性质,所以在负载药物和DNA方面已显示出优良的应用前景[12]。Zhang 等[13]将GQDs用于干细胞标记,发现GQDs 比较容易进入细胞,且表现出较低的细胞毒性,可以产生清晰稳定的图像。利用GQDs可以与多种分子结合的性质,Wang等[14]用粒度为15 nm 的PEG 对GQDs进行表面修饰,利用氢键将阿霉素(DOX)结合到GQDs 表面形成DOX@GQDs⁃PEG。GQDs⁃PEG 作为药物载体,运载能力可以达到2.5 mg/mg,可以通过调节pH 值控制其对药物的释放。Sui等[15]发现GQDs与顺铂(CDDP)结合后,由顺铂导致的细胞毒性增大,细胞周期停滞以及DNA 分裂程度加强。GQDs 通过提高细胞通透性来增加其细胞摄取量,在细胞内增强其与DNA 的相互作用,从而改善CDDP的药物性能。这说明GDQs可以通过增强细胞摄取药物能力来提高抗肿瘤药物的化疗效果。
为了提高DNAzyme 在细胞内的稳定性,选用两个靶向TNF⁃α基因的DNAzyme与GQDs 结合。虽然目前已有多种抑制TNF⁃α 蛋白表达的方法,包括利用靶向TNF⁃α 的siRNA[16],但是siRNA 的转入一般比较繁琐[17],而且由于siRNA 的稳定性差,所以最终效率比较低。本文系统研究以GQDs 为载体的复合体系在细胞水平的稳定性以及它们抑制TNF⁃α 蛋白表达的能力。
1 材料与方法
1.1 材料和试剂
人宫颈癌细胞系Hela 和人乳腺癌细胞MCF⁃7 细胞均购于中科院上海细胞库。培育Hela 使用的DMEM 培养基和培养MCF⁃7使用的RPMI⁃1640培养基均补充添加10%加强型新生牛血清以及1%青霉素⁃链霉素双抗溶液。细胞于37 ℃含5%CO2的培养箱中培养。DMEM 培养基和RPMI1640 培养基购自赛默飞世尔科技有限公司;胎牛血清购自美国Gibco 公司;青霉素链霉素双抗溶液和Hoechst 33258 购自上海碧云天生物科技有限公司。BCA蛋白定量试剂盒购自北京康为世纪生物科技有限公司;ECL发光试剂盒购自Solar⁃bio公司。GAPDH和二抗(goat⁃anti⁃rabbit)购自Protein⁃tech公司。GQDs由上海交通大学微纳米科学科技研究院郭守武教授课题组提供。实验所用核苷酸序列是根据文献设计的[20⁃21],均由上海生工生物工程有限公司合成,经UltraPAGE纯化。具体序列如下:
TNF⁃DNAzyme1:5’⁃FAM⁃GTGCTCAGGCTAGCTAC AACGAGGTGTCC⁃dT⁃3'
TNF⁃DNAzyme2:5'⁃FAM⁃GCAGAAGAGGGCTAGCTAC AACGAGTGGTGGCG⁃dT⁃3'
Substrate1:5'⁃GGACACCrArUGAGCAC⁃3'
Substrate2:5'⁃CGCCACCACrArUTCTTCTGC⁃3'
Cu⁃DNAzyme:5'⁃TGAGTGAGTCTGGGCCTCTTTTTAA GAAC⁃3'
Substrate for Cu⁃DNAzyme:5'⁃GAATTCTAATACGACTC AGCCG⁃FAM⁃3'
1.2 测试和表征
荧光分光光度计(中国Angilent Technologies 公司Cary Eclipse);化学发光仪(中国天能公司Tanon 5200S);垂直电泳槽(中国北京六一仪器厂Mini⁃Protean Tetra);酶标仪(美国Biotek公司Synergy 2)。
1.3 实验步骤
1.3.1 GQDs和单双链DNA(sDNA/dsDNA)的相互作用
末端荧光标记的sDNA 退火后,与一定浓度的GQDs 混合均匀,通过检测体系荧光强度变化来考察GQDs 和sDNA 的相互作用。同样,通过检测部分互补配对的DNAzyme⁃底物体系与不同浓度的GQDs相互作用后的荧光强度来考察GQDs 与双链DNA 的相互作用。为了验证被GQDs 吸附之后的DNAzyme 仍具有活性,以课题组前期研究的铜离子依赖性DNAzyme 为对象进行实验[22]。铜离子依赖性DNAzyme(E)和荧光标记底物(S)1∶1混合后高温退火,加入GQDs和28 μmol/L辅助因子,37 ℃恒温水浴反应30 min,用荧光分光光度计(激发波长,495 nm)检测产物的荧光强度。
1.3.2 TNF⁃DNAzyme体外活性检测
变性聚丙烯酰胺凝胶电泳检测TNF⁃DNAzyme1 的体外活性。若TNF⁃DNAzyme1 具有活性,其能够在金属辅助因子Mg2+存在下有效切割底物。TNF⁃DNA⁃zyme1/Substrate1 复合体系高温退火后配置20 μL 的反应体系(含2 μL 20 μmol/L 的酶和底物复合溶液,16 μL Tris⁃HCl 溶液,2 μL 200 mmol/L Mg2+),37 ℃恒温反应30 min;反应结束后加入4 μL 电泳上样缓冲液(8 mol/L 尿素,0.3 mol/L 蔗糖,50 mmol/L 乙二胺四乙酸二钠,5 mmol/L Tris⁃硼酸溶液,pH 8.3),混合均匀,离心,80 ℃金属浴中加热5 min 后立即放入冰盒,等待上样;28%聚丙烯酰胺凝胶电泳分离DNA 序列。底物切割效率计算公式:P=1-Sr/St,其中Sr代表底物剩余的量,St代表反应前底物的总量。
1.3.3 细胞中总蛋白的提取
Hela细胞以4×105mL-1的密度接种于6孔板中,贴壁后用含有20 mmol/L Mg2+的培养基预处理60 min,PBS 充分洗涤,加入2 mL 含有TNF⁃DNAzyme1/GQDs的培养基。其中GQDs 浓度为50 μg/mL,TNF⁃DNA⁃zyme1 浓度为40 nmol/L。细胞在细胞培养箱中分别培育0、12、24、36 和48 h。对于含有TNF⁃DNAzyme2,或者GQDs 等体系实验操作同上。细胞培育结束后,用细胞裂解液裂解细胞,提取总蛋白。
1.3.4 BCA试剂盒检测蛋白浓度
800 μL 蛋白标准配制液加入到蛋白标准液(20 mg BSA)中,充分混合,配成25 mg/mL 的蛋白标准溶液;取蛋白标准液适量,稀释至浓度为5 mg/mL;按体积比50∶1 混合BCA 试剂盒A 液B 液制成BCA 工作液;将标准品按0、1、2、4、8、12 和16 μL 加到96 孔板的标准品孔中,每个孔用标准品稀释液补足至20 μL;用标准品稀释液将待测蛋白稀释至适当浓度,取20 μL 样品,加到96 孔板的样品孔中;每孔加入200 μL BCA 工作液,37 ℃反应30 min;酶标仪检测样品在562 nm 处的吸收值,根据标准曲线确定样品的蛋白浓度。
1.3.5 Western Blot
Western Blot 检测Hela 细胞中TNF⁃a 蛋白的表达水平。蛋白上样量为10 μg,恒定电压80 V 经分离胶电脉20 min 后,调整电压为120 V,直至上样缓冲液中的蓝色条带到达胶的底部;用200 mA 恒定电流转膜;TBST洗涤PVDF膜4×10 min;PVDF膜用5%的脱脂牛奶封闭1 h;4 ℃下用一抗孵育过夜;TBST 洗涤2×10 min;再用二抗常温孵育1 h。利用超敏ECL 化学发光检测试剂盒检测结果。
1.3.6 细胞毒性检测
取对数生长期Hela 细胞接种于96 孔板,每孔100 μL,常规培养12 h 后加入含20 mmol/L Mg2+的培养基预处理60 min;结束后用PBS 清洗2 次;细胞中加入分别含有TNF⁃DNAzyme1、TNF⁃DNAzyme2、TNF⁃DNAzyme1/GQDs、TNF⁃DNAzyme2/GQDs 和GQDs的同体积培养基,在37 ℃、5 % CO2、饱和湿度下培育36 h。每组设5个平行复孔,另设相同平行复孔的空白组、对照组。结束培育后,PBS 充分洗涤,加入100 μL MTT 继续培养4 h;加入150 μL DMSO,轻微摇荡10 min,酶标仪检测细胞在570 nm处的吸光度值A。细胞存活率(%)=(实验组A值/对照组A值)×100%。
2 结果和讨论
2.1 GQDs的表征
实验用的GQDs是单层石墨烯量子点,其荧光强度较弱,在细胞中很难直接观察到其荧光[18]。为了确认作为载体GQDs 能否进入细胞,利用GQDs 可以淬灭小分子的荧光的特性,首先将细胞用Hoechst染色,细胞核呈现蓝色Hoechst荧光;利用GQDs可以有效淬灭小分子荧光的特点,将染色后的细胞与GQDs共培育,跟踪GQDs是否进入细胞。如图1所示,随着GQDs浓度的增加,共培育时间的增长,细胞核内的Hoechst可以被GQDs完全淬灭,表明GQDs能够进入到细胞核[19]。因此,GQDs具备作为DNAzyme载体的基本能力。
2.2 DNAzyme/GQDs体系的构建
利用6'⁃FAM 标记的单双链DNA 的荧光强度变化来检测GQDs 负载单双链DNA 的能力[23]。如图2(a)所示,随着GQDs 浓度的增加,sDNA 的荧光强度随之降低,当GQDs浓度达到200 μg/mL时,体系中荧光强度基本为零。从图2(b)可以看出,随着体系中GQDs浓度不断增加,检测到DNAzyme⁃底物的双链DNA(ES)的荧光强度越来越低。当GQDs浓度达到500 μg/mL,体系中的荧光强度基本为零,说明此时的ES 和GQDs 完全结合。由此可以初步确定,GQDs 能够负载单链DNA以及DNAzyme⁃底物体系。
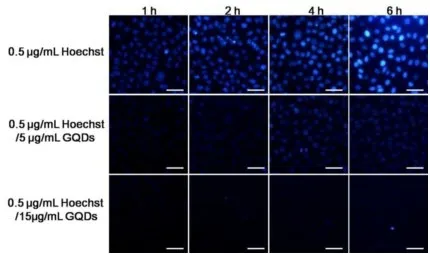
图1 MCF⁃7细胞与Hoechst和Hoechst/GQDs分别共培育不同时间得到的荧光图Figure 1 Fluorescence images of the MCF⁃7 cells after incubation with Hoechst and Hoechst/GQDs for different time
利用课题组前期研究的铜离子依赖性DNAzyme(Cu⁃DNAzyme),考察负载在GQDs 上DNAzyme 的活性[24]。如图2(c)所示,GQDs 和ES 复合体系中加入辅助因子,37 ℃恒温反应30 min 后,仅检测到微弱的荧光;但在体系中加入不同浓度的乙醇溶液后,体系的荧光信号增强。当加入50%的乙醇时,荧光强度基本接近自由的ES 的荧光强度。由图2(d)可知,被GQDs 吸附的ES体系在铜配合物存在下仍能表现出活性,但由于产物被GQDs 吸附,检测到的荧光强度较低,当有乙醇存在时,减弱了sDNA和GQDs之间的疏水作用,使得带有荧光标记的sDNA 游离到溶液中,荧光强度增加。为了确认检测到的荧光是来自荧光标记的DNA 产物,在带有荧光标记的底物中加入足量的GQDs,后加入50%的乙醇溶液,充分混合后发现体系没有荧光。同样,ES体系也基本未被检测到任何荧光信号。
2.3 TNF⁃DNAzyme1的体外活性
根据文献[20],以具有切割磷酸酯键能力的10⁃23 DNAzyme 为模版,设计合成了TNF⁃DNAzym1 和TNF⁃DNAzym2,具体见图3(a)和3(b)。两个DNA⁃zyme 具有和10⁃23 DNAzyme 相似的辅助因子镁离子。首先考察镁离子浓度对TNF⁃DNAzyme1 活性的影响,结果如图3(c)所示。由图3(d)可知:当保持TNF⁃DNAzym1 和底物浓度保持不变时,随着镁离子浓度增加,TNF⁃DNAzyme1 切割活性逐渐增加。当镁离子浓度为50 mmol/L 时,切割效率高达60 %左右,且保持在稳定的水平。在此条件下,反应时间为30 min 时,TNF⁃DNAzyme1 活性达到最高,大约70 %的底物得到切割。TNF⁃DNAzyme1 活性与体系的pH 值也是相关的。由图3(e)可知:在中性或偏碱性的环境中,TNF⁃DNAzyme1 活性达到最高;TNF⁃DNAzym2 的结构与TNF⁃DNAzyme1 相似,但其在分子水平活性很低,有可能是因为它的稳定性较差。
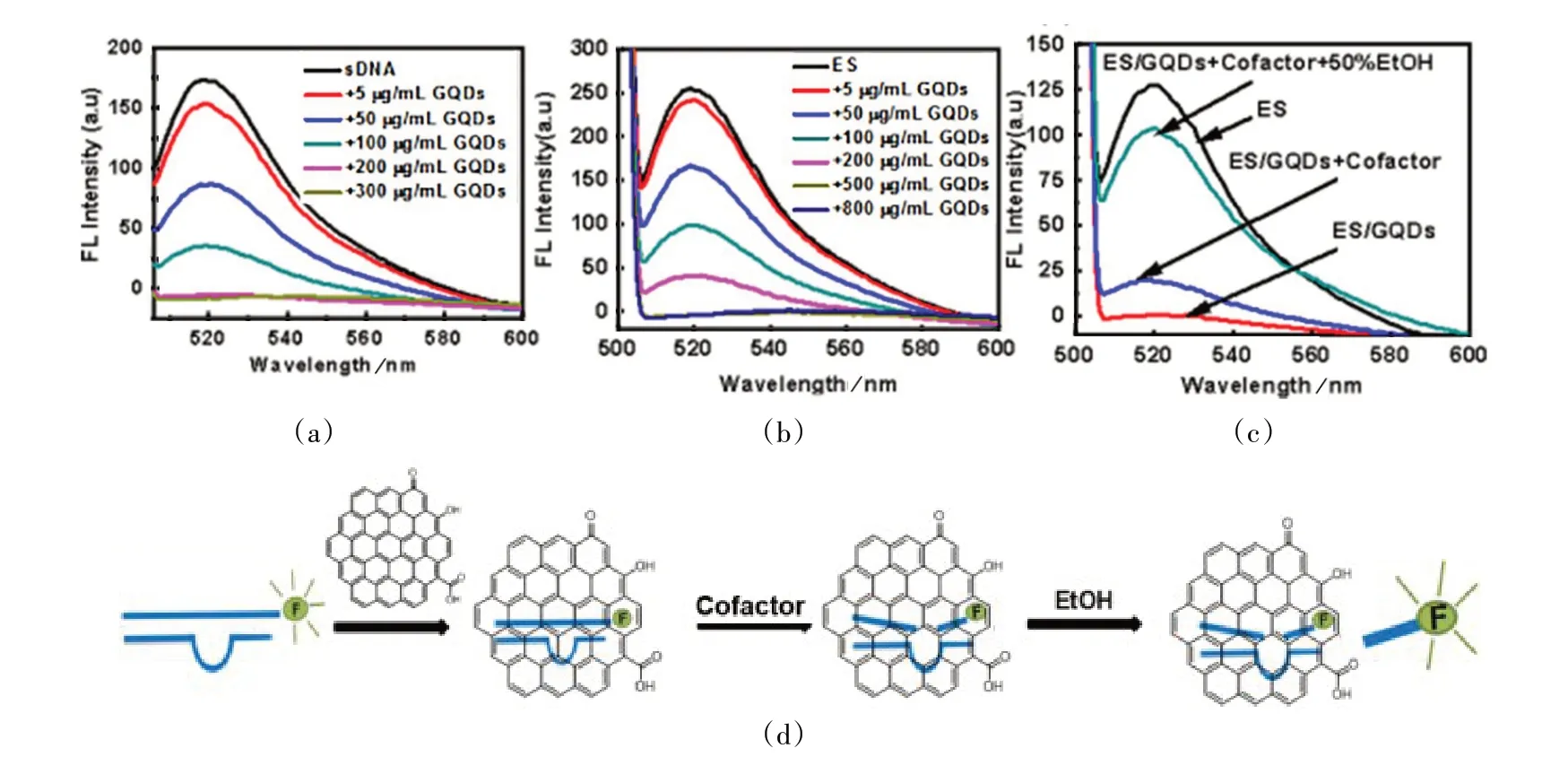
图2 荧光光谱法检测GQDs和单链DNA(sDNA)Figure 2 The interaction between GQDs and sDNA and ES detected by the fluorescence change of the system
2.4 TNF⁃DNAzyme1/2对TNF⁃α蛋白表达的影响
TNF⁃DNAzyme1 或TNF⁃DNAzyme2 单独与Hela 细胞作用48 h 后,细胞中TNF⁃a 蛋白的表达没有明显变化,结果如图4(a)和4(b)所示。而以GQDs 为载体将TNF⁃DNAzyme1 导入到细胞后发现,随着TNF⁃DNA⁃zyme1/GQDs 作用时间的加长,Hela 细胞内TNF⁃α 蛋白的表达量逐渐下降。当作用36 h 后,TNF⁃DNA⁃zyme1/GQDs 体系对细胞的影响达到最大,蛋白的表达量降低了35%,见图4(c)。
TNF⁃DNAzyme2/GQDs体系对TNF⁃α蛋白在细胞水平的表达影响更加显著。如图4(d)所示,随着TNF⁃DNAzyme2/GQDs 与细胞共培育时间增长,TNF⁃α 蛋白的表达量明显下降。当TNF⁃DNAzyme2/GQDs 与细胞作用36 h 后,细胞内TNF⁃α 蛋白的表达量降低了近50%。值得注意的是,在同等条件下GQDs 单独与细胞共培育后,TNF⁃α 蛋白表达量随GQDs 浓度的升高而增大。当GQDs 浓度为100 μg/mL 时,蛋白表达量达到最大,见图4(g)。虽然目前还不知道GQDs 提高TNF⁃α表达的机理,但是可以肯定的是,这一结果表明TNF⁃DNAzyme/GQDs 体系对TNF⁃α 蛋白表达的影响应该比实验观测到的更为显著。
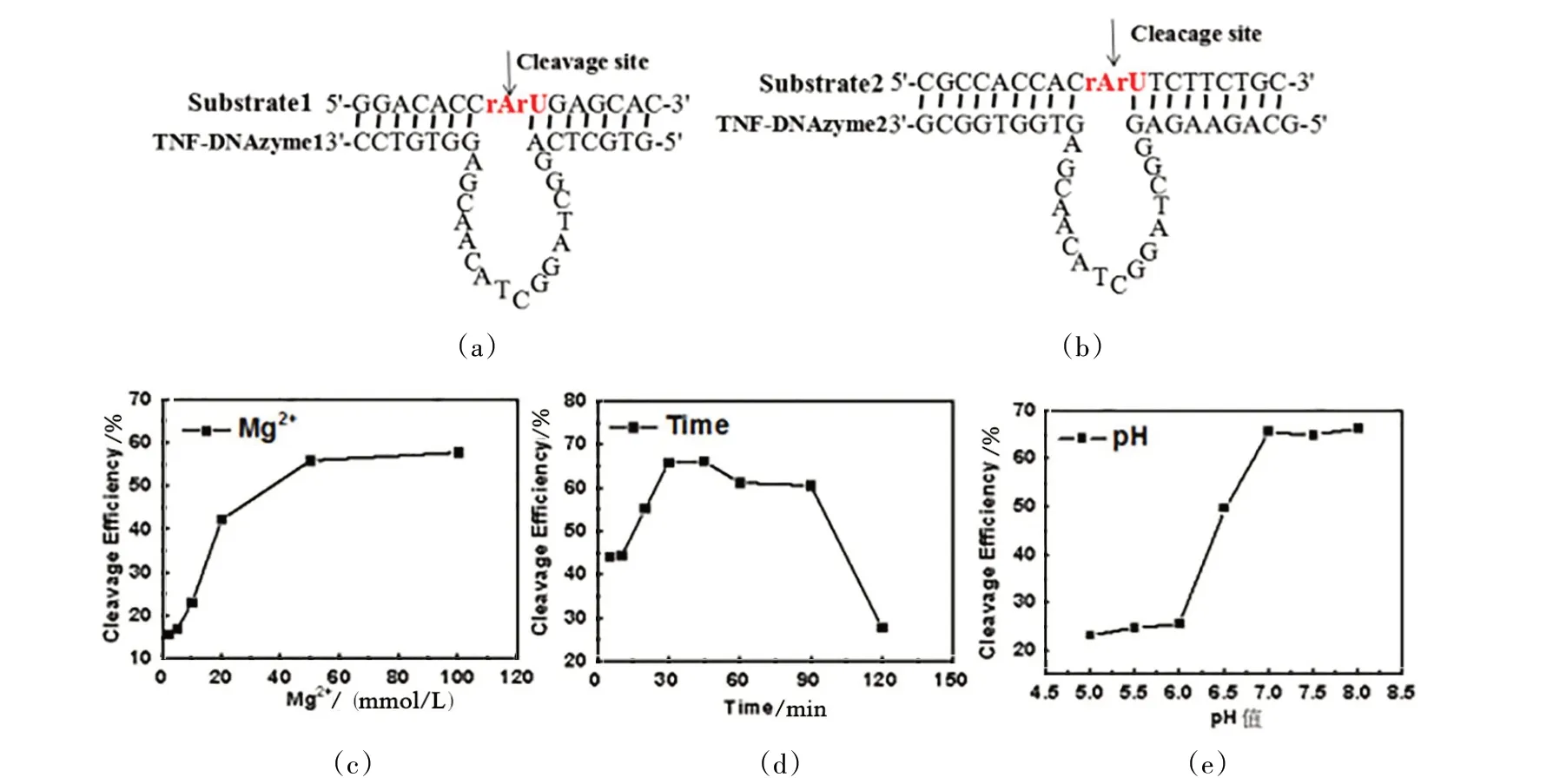
图3 镁离子浓度、反应时间和pH值对TNF⁃DNAzyme1活性的影响Figure 3 Effects of Mg2+concentration,reaction time and pH on the TNF⁃DNAzyme1 activity
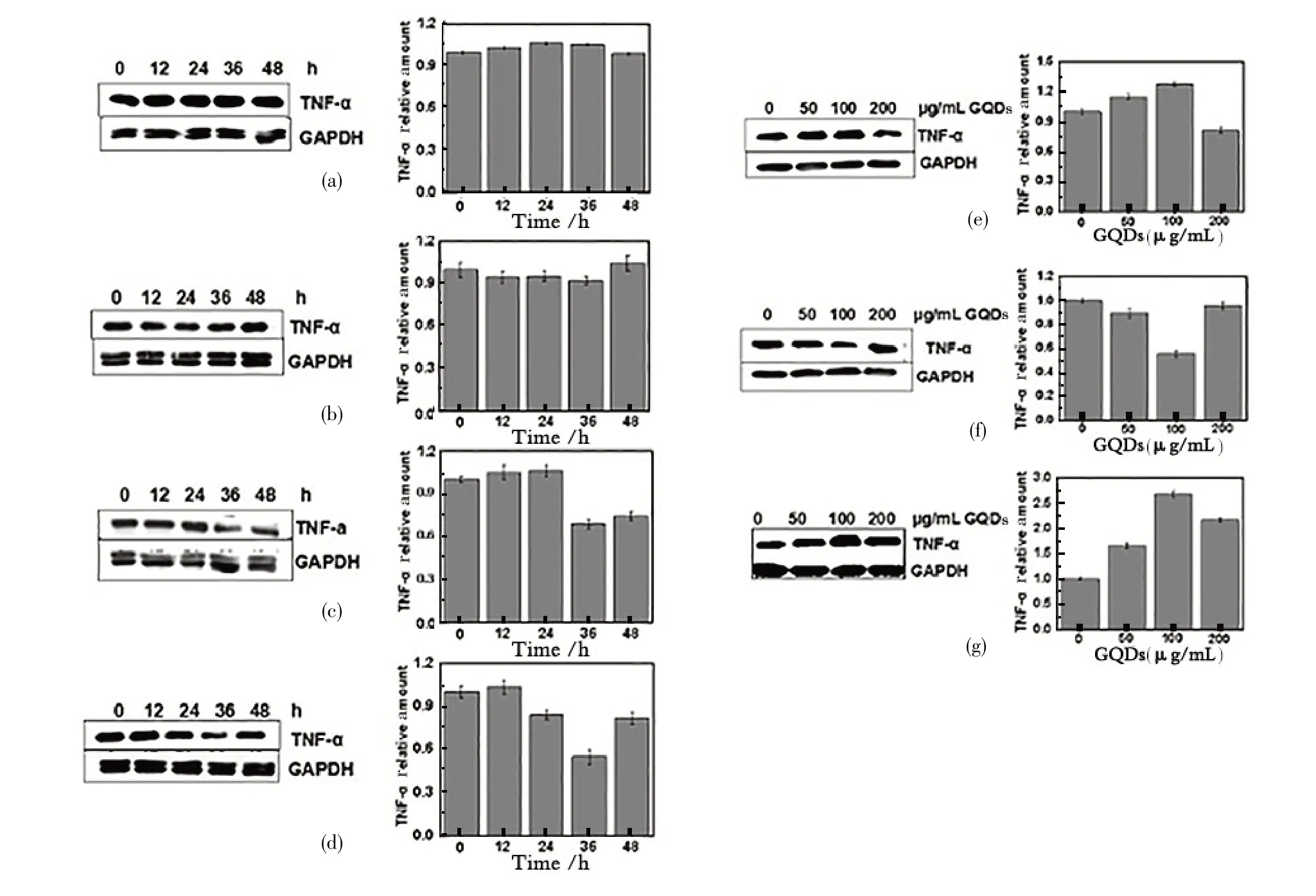
图4 TNF⁃DNAzyme1/2对TNF⁃α蛋白表达的影响Figure 4 Effects of TNF⁃DNAzyme1/2 on the expression of TNF⁃α
为进一步确认和优化TNF⁃DNAzyme1/GQDs 和TNF⁃DNAzyme2/GQDs 体系对TNF⁃α 蛋白表达的影响,考察了不同浓度GQDs 对TNF⁃DNAzyme1 和TNF⁃DNAzyme2 活性的影响。从图4(e)可以看出:增加GQDs 浓度的TNF⁃DNAzyme1 对细胞内TNF⁃α 蛋白表达量只是略有影响;而TNF⁃DNAzyme2/GQDs 作用的细胞的TNF⁃α蛋白表达量随GQDs浓度升高明显降低,当GQDs 浓度为100 μg/mL 时,TNF⁃α 蛋白表达量达到最低,只有原来的50%左右。GQDs浓度在200 μg/mL时,目的蛋白的表达量反而有小幅度的上升,这可能是高浓度的GQDs和细胞培育36 h后造成细胞凋亡,见图4(f)。基于和单独TNF⁃DNAzyme,单独GQDs 与细胞作用的结果比较,TNF⁃DNAzyme 的活性在GQDs 存在的条件下明显得到了提高。与体外分子水平的结果对照,推测TNF⁃DNAzyme 活性提高的可能原因是其稳定性得到了改善。当然TNF⁃DNAzyme在细胞内的状态和体外有可能不同,因而作用机理等方面也有可能有差异;此外,因为GQDs的协助TNF⁃DNAzyme也可能在细胞内的累积量增加,因此也可能导致其活性提高。这些推断还有待于进一步实验验证。
2.5 TNF⁃DNAzyme/GQDs体系的细胞毒性
为进一步了解该体系的作用效果和实际应用的可能性,分别检测了TNF⁃DNAzyme1、TNF⁃DNAzyme2、GQDs、TNF⁃DNAzyme1/GQDs 以及TNF⁃DNAzyme2/GQDs 体系对细胞活力的影响,结果(图5)显示,与TNF⁃DNAzyme1、TNF⁃DNAzyme2 单独培育36 h 的Hela细胞,细胞存活率受TNF⁃DNAzyme1和TNF⁃DNAzyme2浓度影响不大,细胞始终保持较高的活力,见图5(a)。与TNF⁃DNAzyme1/GQDs 和TNF⁃DNAzyme2/GQDs作用的细胞,存活率受GQDs 浓度影响显著,随浓度的升高而下降,在大于100 μg/mL 时,细胞凋亡明显,存活率低于50%。Mg2+的预处理对细胞存活率没有明显的影响。TNF⁃DNAzyme/GQDs 体系的毒性主要来源于GQDs,但只要使用较低浓度的GQDs,体系的细胞毒性不影响DNAzyme的活性测定。
3 结论

图5 TNF⁃DNAzyme/GQDs体系的细胞存活率Figure 5 Cell viability of Hela cells after treating with different concentrations of TNF⁃DNAzyme/GQDs
为了提高DNAzyme 细胞水平的稳定性,设计了两个靶向TNF⁃α 的DNAzyme,TNF⁃DNAzyme1 和TNF⁃DNAzyme2,探索它们和GQDs 形成复合体系时的稳定性以及在细胞水平的活性。研究发现,TNF⁃DNA⁃zyme1、TNF⁃DNAzyme2 单独与Hela 细胞作用不会对TNF⁃α 的蛋白表达水平产生显著影响,但是当TNF⁃DNAzyme与一定浓度的GQDs结合后,TNF⁃DNAzyme1/GQDs 和TNF⁃DNAzyme2/GQDs 体系均能显著降低Hela 细胞内TNF⁃α 蛋白的表达量,而且这些降低的结果是在GQDs 本身增加Hela 细胞内TNF⁃α 蛋白的表达量的基础上,说明实际降低量比观测到的更高。此结果表明GQDs 可以提高TNF⁃DNAzyme 在细胞内的活性,从而抑制目的蛋白表达。基于对照实验以及体外分子水平的实验,推测DNAzyme 在细胞水平的高活性可能是因为GQDs 提高了它的稳定性。当然DNAzyme的稳定性是否直接导致它的活性增强,还需要其他机理实验来验证,但是这一初步结果表明利用DNAzyme调控蛋白表达的可能性。
