高压下Mg3P2晶体结构的相变与物性研究
2021-04-27刘涵张奥刘思远程宇衡刘艳辉
刘涵, 张奥, 刘思远, 程宇衡, 刘艳辉
( 延边大学 理学院,吉林 延吉 133002 )
0 引言
由于第Ⅱ主族碱土金属元素与第Ⅴ主族元素形成的A3B2型化合物具有较宽的带隙,因此该型化合物成为制备电子和光电子器件的良好材料[1].近年来,因Mg3X2(X=N,P,As,Sb)具有良好的热电性质[2]和拓扑性[3]而受到研究者们的广泛关注.氮化镁(Mg3N2)粉末是一种常用的固体催化剂,目前被广泛应用于合金、陶瓷和储氢材料等合成方面,以及催化聚合物的交联反应中[4-7].2012年,Song等[8]研究显示,金属Mg与P直接反应可生成一种新型的三维Mg3P2树突结构,同时Song等还对一种具有可调形状的Mg3P2微结构的生长机理进行了阐述.Xia等[9-10]的研究表明Mg3Sb2具有良好的热电性质,且静水压力能够提高n型和p型Mg3Sb2的热电性能.另外,研究还显示离子掺杂剂[11-12]和双轴应变工程[13-16]等对n型Mg3Sb2的热电性能具有良好的改善作用.Kazuki等[17]研究表明,消除晶界电阻可使晶体的ZT值在室温下提高两倍以上.由于A3B2型化合物在常压下的各种优异性能,其在高压下的性质也引起了研究者们的关注.2009年,Rebecca等[18]对Mg3N2的高压行为进行预测显示,Mg3N2在0~100 GPa压力范围内发生了两次相变,相变序列为Ia-3(0 GPa)→C2/m(22 GPa)→P-3m1(65 GPa),且这3相均为直接带隙半导体,带隙分别为1.65 eV(0 GPa)、2.35 eV(25 GPa)和3.45 eV(70 GPa),即带隙随着压强的增大而增大.2019年,Yang等[1]对Mg3As2的高压行为进行预测显示,Mg3As2的相变序列为Ia-3(0 GPa)→P-3m1(1.3 GPa)→C2/m(12 GPa)→P-1(30 GPa),其中Ia-3和P-3m1相为直接带隙半导体,而C2/m和P-1相则转变为间接带隙半导体.该研究结果与文献[18]的结果有所不同的是,在高压下Mg3As2相的带隙随着压强的增大而减小.目前,关于Mg3P2在高压下的性质研究得较少,为此本文基于密度泛函理论的第一性原理并结合晶体结构预测技术,在0~100 GPa压力范围内,研究了Mg3P2的相变序列,并且对其电子性质和成键行为进行了研究.
1 计算方法
使用基于粒子群优化算法的CALYPSO软件[19]预测晶体结构,预测的压力范围为0~100 GPa,其中模拟晶胞为2倍胞和4倍胞.运用VASP软件包[20]对所预测的结构进行优化,并计算其能带及其电子结构.在计算参数设置上,描述电子之间的交换关联势采用广义梯度近似(Generalized Gradient Approximation,GGA)下的Perdew-Burke-Ernzerh(PBE)交换关联泛函[21].Mg和P原子的价电子分别为2p63s2和3s23p3.为使Mg3P2的能量收敛于1 meV/atom,对Mg3P2平面波的截断能进行了能量收敛测试,为700 eV.采用Monkhorst-Pack网格方法计算第一布里渊区内的积分[22],网格间距为0.25 nm-1.在空间群不改变的情况下,优化晶胞参数和原子位置,自洽能量收敛设置为0.001 eV/Å.应用PHONOPY软件计算声子色散关系[23].
2 结果与讨论
2.1 高压下预测晶体结构的相变
在0~100 GPa压力范围内,应用CALYPSO晶体结构搜索技术得到了Mg3P2晶体结构的相变序列.对预测的Mg3P2晶体结构进行优化时,由于本文在计算时不考虑温度,即T=0 K,因此根据公式G=H-TS(其中G为吉布斯自由能,H为焓,T为温度,S为熵)可知,可以用焓代替自由能.晶体结构优化结果如图1所示.由图1可以看出:在0~100 GPa范围内,Mg3P2晶体的相变序列为Ia-3→P-3m1→C2/m→Cmc21.常压下,空间群为Ia-3的Mg3P2晶体结构的焓值最低,该结果与文献[24]的结果一致;当压力达到2.5 GPa时,Ia-3结构转变为P-3m1结构;当压力为19.9 GPa时,P-3m1结构转变为C2/m结构;当压力为48.2 GPa时,C2/m结构转变为Cmc21结构.在文献[24]中,作者还报道了在0~100 GPa压力范围内,Mg3P2晶体结构还存在C2/c结构和P63/mmc结构,但这两项结构并未出现在本文的预测结果中.为了进一步确定Mg3P2晶体在高压下能够稳定存在的结构,本文在图1中用虚线给出了文献[24]报道的这两个结构.由图1中的虚线可以看出,这两个结构在焓差曲线图中具有较高的能量,由此可知这两个结构在高压下不能稳定存在.对Mg3P2晶体的体积与压力的关系进行计算显示:当压力达到19.9 GPa时,P-3m1相转变为C2/m相的体积坍塌率为4.67%;当压力达到48.2 GPa时,C2/m相转变为Cmc21相的坍塌率为1.96%,均属于一级相变.
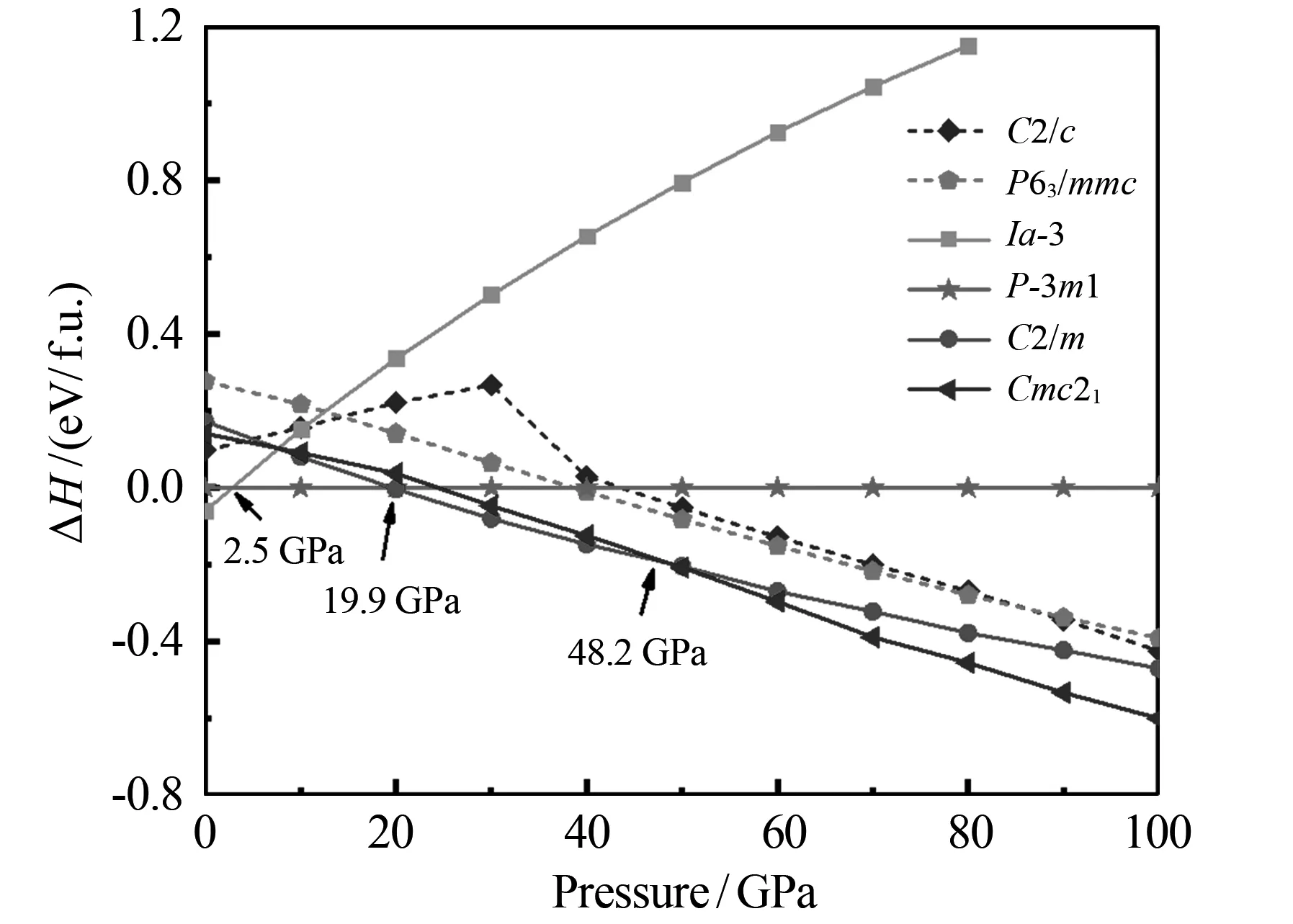
图1 预测得出的Mg3P2晶体结构的焓差曲线
表1给出了Mg3P2高压结构的平衡态晶格常数和原子位置,预测得出的3个高压相的晶体结构如图2所示.在P-3m1相中,优化的晶格常数是a=b=4.163 Å,c=6.581 Å,α=β=90.000°,γ=120.000°.Mg原子的Wyckoff占位是2d(-0.667,0.667,0.644)和1a(0.000,0.000,0.000),P原子的Wyckoff占位是2d(-0.667,0.667,0.235).Mg原子与最近邻的P原子之间的距离为2.532 Å.Mg原子与P原子存在2种结合方式,分别是1个Mg原子与6个P原子连接形成八面体结构以及1个Mg原子与4个P原子连接形成三棱锥的四面体结构.在C2/m相中,优化的晶格常数是a=13.308 Å,b=3.742 Å,c=6.896 Å,α=γ=90.000°,β=117.842°.Mg原子的Wyckoff占位是2a(0.000,0.000,0.000)、2d(0.000,0.500,0.500)、4i(0.843,0.500,0.020)和4i(0.250,0.500,0.662),P原子的Wyckoff占位是4i(0.895,0.500,0.727)和4i(0.133,0.000,0.746).Mg原子与最近邻的P原子之间的距离为2.393 Å.Mg原子与P原子存在3种结合方式,分别是1个Mg原子与6个P原子连接形成八面体结构,1个Mg原子与4个P原子连接形成三棱锥的四面体结构以及1个Mg原子与4个P原子连接形成平面结构.在Cmc21相中,优化的晶格常数是a=3.852 Å,b=13.120 Å,c=4.988 Å,α=β=γ=90.000°.Mg原子的Wyckoff占位是4a(0.000,0.803,0.676)、4a(0.000,0.993,0.965)和4a(0.500,0.897,0.233),P原子的Wyckoff占位是4a(0.000,0.188,0.679)和4a(0.500,0.921,0.716).Mg原子与最近邻的P原子之间的距离为2.246 Å.Mg原子与P原子存在2种结合方式,分别是1个Mg原子与6个P原子连接形成八面体结构以及1个Mg原子与5个P原子连接形成六面体结构.

表1 P-3m1相、C2/m相和Cmc 21相的晶格参数和原子位置

(a) P-3m1相(2.5 GPa) (b) C2/m相(19.9 GPa) (c) Cmc21相(48.2 GPa)图2 高压下Mg3P2的3种晶体结构
2.2 高压下Mg3P2晶体结构的稳定性
研究表明,晶体结构若具有动力学稳定性,其在布里渊区内所有的声子振动频率应为正值[25-26].为了确定预测得到的Mg3P2晶体高压结构是否具有动力学稳定性,分别计算了3个高压相的声子谱,结果如图3所示.图3(a)、图3(b)和图3(c)分别为P-3m1相在2.5 GPa时、C2/m相在19.9 GPa时和Cmc21相在48.2 GPa时的声子色散关系和声子态密度(PHDOS).通过声子谱分析可以发现,在各自的布里渊区内,3个相均未出现虚频,由此表明本文预测的高压相均具有动力学稳定性.
2.3 高压下Mg3P2晶体结构的电子性质
为了研究各高压相的电子性质,计算了Mg3P2高压相的能带结构和电子态密度.图4(a)、图4(b)和图4(c)分别为P-3m1相、C2/m相和Cmc21相晶体结构的电子能带结构和态密度图.由图4可以看出,3个相的导带底部和价带顶部都没有穿越费米面,其带隙分别为1.22 eV、0.74 eV 和0.04 eV,其中P-3m1相和C2/m相为间接带隙半导体,Cmc21相为直接带隙半导体.对各相的电子态密度进行分析表明,费米面附近的态密度主要由P原子的p轨道贡献.

(a) P-3m1相(2.5 GPa) (b) C2/m相(19.9 GPa) (c) Cmc 21相(48.2 GPa)图3 Mg3P2高压结构中各相的声子色散关系与投影态密度
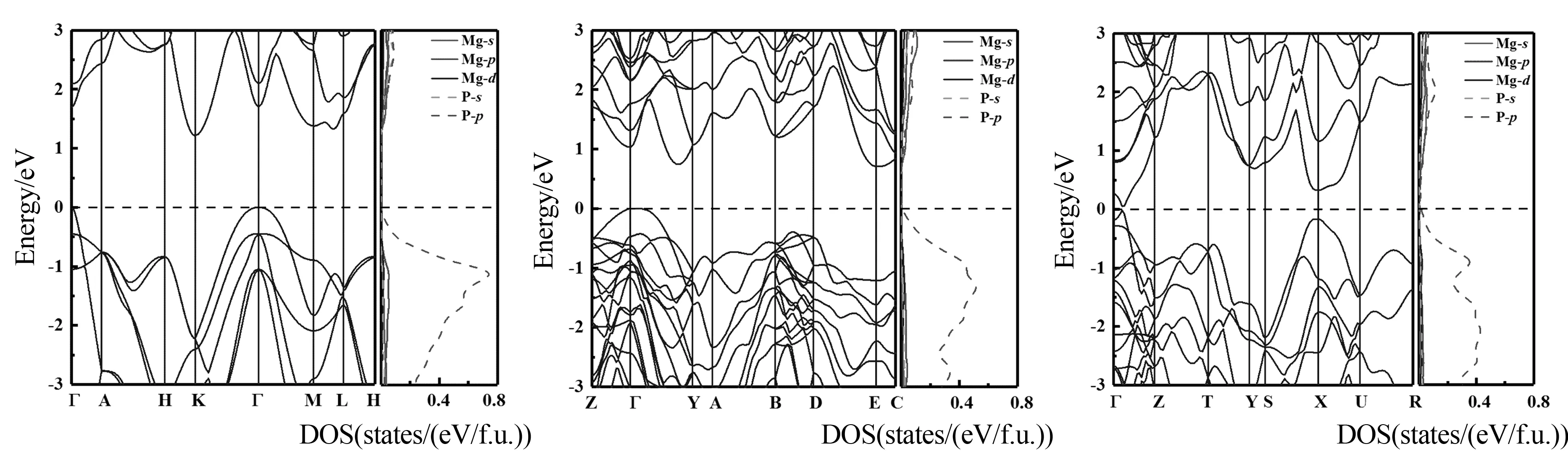
(a) P-3m1相(2.5 GPa) (b) C2/m相(19.9 GPa) (c) Cmc 21相(48.2 GPa)图4 Mg3P2高压结构中各相的能带结构和电子态密度
为确定Mg3P2高压相晶体结构的化学键,计算了P-3m1相、C2/m相和Cmc21相的电子局域函数,并绘制了二维的电子局域函数图,如图5所示.图5中3个相的等值面分别选取的是0.8、0.8和0.85.由图5可以看出,电子局域明显存在于P原子周围,且每个P原子周围存在Mg原子贡献的局域电子.该结果表明,高压下Mg和P原子均以极性共价键存在.
为了了解Mg3P2晶体结构的电子特性,对其Bader电荷转移[27-28]进行了计算,结果如表2所示.表2中的数据显示:P原子的电负性强于Mg原子,P原子是受主,Mg原子是施主;在P-3m1相中平均每个Mg原子失去0.82 e,每个P原子得到1.23 e;在C2/m相中平均每个Mg原子失去0.77 e,每个P原子得到1.15 e;在Cmc21相中平均每个Mg原子失去0.77 e,每个P原子得到1.16 e.上述结果表明,随着压力的增加Mg失去电子的能力逐渐减弱.

(a) P-3m1相(2.5 GPa) (b) C2/m相(19.9 GPa) (c) Cmc 21相(48.2 GPa) 图5 高压下Mg3P2结构中各相的电子局域函数
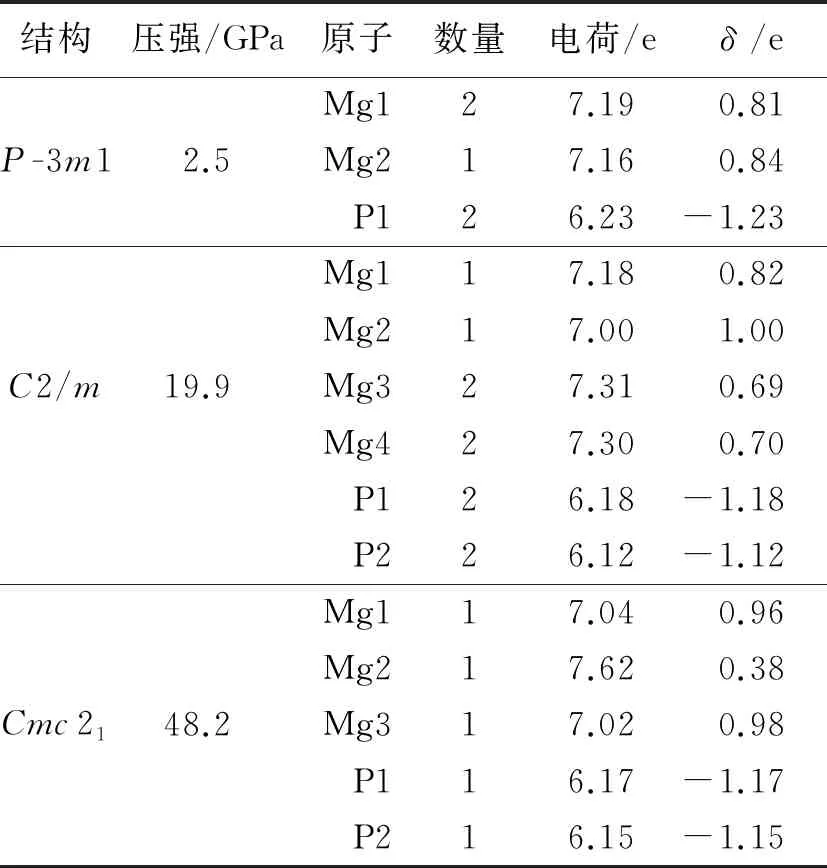
表2 Mg3P2高压结构中各相的Bader电荷转移
3 结论
本文基于第一性原理并结合卡利普索晶体结构预测方法在0~100 GPa范围内对Mg3P2晶体的相变行为进行预测显示,Mg3P2晶体结构相变序列为Ia-3→P-3m1→C2/m→Cmc21.对Mg3P2晶体的声子色散关系进行分析显示,3个高压相具有动力学稳定性.对Mg3P2晶体的能带结构进行计算显示,3个高压相均呈现半导体性质,其中P-3m1相与C2/m相为间接带隙半导体,Cmc21相为直接带隙半导体.对Mg3P2晶体的电子态密度进行计算显示,费米面附近的态密度主要由P原子的p轨道贡献.对Mg3P2晶体的电子局域函数进行计算显示,电子局域围绕在P原子周围,说明Mg原子和P原子之间形成的是极性共价键.对Mg3P2晶体的Bader电荷转移进行计算显示,P原子具有较强的电负性,且电荷在压力作用下由Mg原子向P原子转移.本文研究结果可为研究Mg3P2的物性提供理论参考.
