腓骨肌萎缩症2A2A型家系的神经电生理及基因突变分析
2021-04-20赵伯杰赵鼎李瑞
赵伯杰,赵鼎,李瑞
遗传性运动和感觉神经病(hereditary motor and sensory neuropathies,HMSN)又称腓骨肌萎缩症(charcot-marie-tooth,CMT),于1886年由法国Jean和Piere及英国医生Howard率先报道,并命名为Charcot-Marie-Tooth病[1],发病率为1/2500[2-3]。主要表现为进行性对称性远端感觉减退,肌无力、肌萎缩、腱反射减弱或消失,以及足部畸形等[4-5]。遗传方式多为常染色体显性遗传,少部分是常染色体隐性遗传、X-性连锁显性遗传和X-性连锁隐性遗传[6]。常染色体显性遗传性CMT依据正中神经传导速度改变分为两型,即脱髓鞘型(CMT1),以神经传导速度减慢(正中神经运动传导速度低于38 m/s),神经活检示显著的髓鞘异常(阶段性脱髓鞘,雪旺细胞增生,呈“洋葱头”样改变)为特征;以及轴索型(CMT2)以神经传导速度正常或轻度减慢(正中神经运动传导速度大于38 m/s),神经活检示慢性轴索变形和再生(轴索变形和有髓纤维减少,神经再生簇形成)为特征[7-9]。迄今已发现的CMT2型致病基因有54个,根据致病基因不同,CMT2A2A被命名为相应亚型。本研究中我们通过对一个典型的CMT2A2A型家系进行临床和基因突变检测分析,旨在加强对该病的认识,为家系遗传咨询及产前诊断提供依据。
1 对象与方法
1.1 研究对象
该CMT汉族家系来自于中国河南省郑州市,3代共7人 (图1)。先证者(Ⅲ代2),男,6岁零10个月,3岁时发现走路不稳,姿势异常,前脚掌无法向上抬起,不会跳。查体示肢体上肢无异常,双下肢远端对称性肌肉无力、肌肉萎缩和感觉障碍,伴跟腱反射消失,出现鹤形腿、高弓足。先证者母亲(Ⅱ代2)查体示双下肢远端对称性肌无力,大腿下1/3肌肉萎缩,呈倒立酒瓶状、末梢型感觉减退、跨阈步态、腱反射消失,并进行过脚部畸形矫正手术。其父亲、姐姐及其他家系成员无相关症状。先证者签署知情同意书后行全外显子组基因测序检测,并对先证者父母、姐姐进行验证。
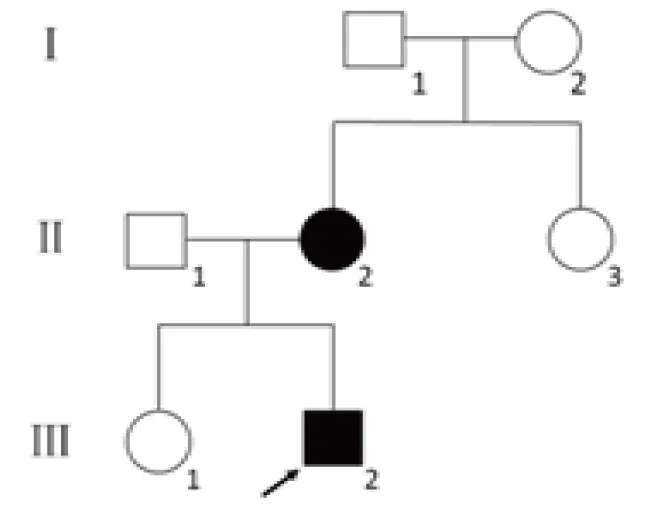
图1 CMT患者系谱图:先证者,○:女性患者,□:男性患者。
1.2 方法
1.2.1 神经电生理检查
对先证者进行周围神经传导和同芯圆针极肌电图(EMG)检测, 采用丹迪Keypoint肌电诱发电位仪检查。运动神经传导及腓肠神经感觉传导检测采用盘状电极记录,鞍形电极刺激,其它感觉传导检测刺激用环状电极(阴阳极间距离应≥2 cm),记录用鞍形电极。带通2~10 kHz,刺激时限0.2 ms,室温21~25 ℃,皮肤温度≥32 ℃。受检患儿安静,手足关节保持放松状态,尺神经运动传导检测时肘关节屈曲20°左右。进行同芯圆针极电极EMG检查时,选择至少3个肢体不同神经支配的4块以上肌肉,同芯圆针电极插入肌肉测定EMG,首先,在肌肉静息状态时,进行多部位探测,观察屏幕上的波形,并监听声音,观察是否有纤颤电位及正锐波等自发电位;然后进行肌肉轻收缩检查,观察并采集屏幕上运动单位电位的波形,测定其平均时限。运动单位电位平均时限与正常值[10]比较,凡时限>20%为时限增宽。
1.2.2 全外显子组基因测序
抽取患儿(先证者)、其姐姐及父母外周静脉血样各2 mL,采用QIAampDNA提取试剂盒(德国QIAGEN公司)提取基因组DNA。然后严格按照Ion-AmpliSeq Exome Kit试剂盒(美国Life公司)扩增目的片段,经过酶切、连接接头序列、纯化再扩增获得基因文库,针对全外显子组基因对先证者及其家系进行全外显子组基因测序。
1.2.3 Sanger测序验证
应用软件Primer Premier 5.0对可疑突变位点的外显子编码区设计引物,应用2×PCR MasterMix聚合酶(上海生工生化科技有限公司),进行PCR扩增(ABI-Veriti96孔梯度PCR仪MFN2上游引物序列为5’-GCTGCCAAGTTGTTTCTGGA-3’,下游引物序列为5’-GGCAGCTCTCTC CACCTATC-3’。PCR反应条件为95 ℃预变性5 min;94 ℃变性30 s;58 ℃退火1 min;72 ℃延伸1 min,进行38个循环,最后72 ℃延伸10 min,25 ℃保存。对PCR产物进行双向直接测序(ABI 3730XL测序仪),结果与参考序列进行比对分析。
1.2.3 突变位点的生物信息学分析
将突变位点在NCBI、HGMD、EXAC、ESP 6 500及千人基因组数据库中进行序列比对分析,以排除该突变位点为已知多态性位点。应用PolyPhen-2和Mutation Taster软件预测突变的有害性及突变位点在物种间的保守性。
2 结果
2.1 神经电生理检查结果
神经传导检查提示先证者感觉和运动传导检测均存在动作电位波幅下降和传导速度减慢,其中胫神经(左)运动传导速度(MCV)为36.2 m/s(正常参考值>42.36 m/s),复合肌肉动作电位(CMAP)波幅为0.12 mV(正常参考值>16.21 mV)[11];胫神经(右)MCV为33.7 m/s,CMAP波幅为1.55 mV(表1、2)。同芯圆针极EMG提示双下肢及左上肢神经及肌肉呈神经原性损害(表3)。

表1 先证者的运动传导测定结果比较

表2 先证者的感觉传导测定结果比较

表3 先证者的同芯圆针极EMG结果比较
2.2 全外显子组基因测序结果
高通量测序显示,先证者1号染色体上的MFN2基因存在第11外显子c.1066A>G错义突变,转录本为NM_014874.4。
2.3 Sanger测序验证结果
Sanger测序结果与MFN2参考序列(NM_014874.4)经比对,发现先证者及其母亲均存在c.1066A>G(p.T356A)错义突变(图2A、B见封二),先证者的这个突变来自于母亲,其父亲与姐姐未携带这个突变位点(图2C、D见封二)。患儿父亲与姐姐未检测到该突变。

2.4 突变位点的生物信息学分析结果
利用生物信息分析软件PolyPhen-2和Mutation Taster对c.1066A>G(p.T356A)突变进行预测,提示其为致病性(图3A、B见封二),MFN2蛋白第356位氨基酸在不同物种间的保守性分析显示突变区域序列在不同物种间高度保守(图3C见封二)。

3 讨论
CMT2A2A型是一种轴突周围神经病变疾病,属于CMT2的一种亚型。该亚型临床表现为四肢远端进行性肌无力及肌肉萎缩,形成“鹤形腿”;足部骨骼畸形,出现高弓足、槌状趾;手部骨间肌出现萎缩,形成爪形手;腱反射减弱或消失;四肢远端感觉受损。少数患者出现脊柱侧凸、视力、听力减退等症状,神经传异速度正常或接近正常。CMT2与CMT1的临床症状类似,但是前者没有后者严重,表现为周围神经没有增大或增厚。CMT是一种高度遗传异质性疾病,即许多不同基因的缺陷可引起相同表型。据国内报道的病例总结,有遗传家族史的占88%,散发病例占12%,与国外报道的相似[12]。CMT2的亚型临床上都比较相似,仅仅分子遗传学上不同。多个研究小组的研究结果表明,MFN2基因突变导致的CMT2A是最常见的CMT2基因型,约占CMT2总数的20%,其临床表现可分为早发肌无力,肌萎缩症状严重和迟发肌无力,肌萎缩症状较轻两种临床表型。MFN2基因热点突变R94Q常导致幼年发病,为重度肌萎缩的轴索型CMT,患者常伴有视神经萎缩的伴随症状[13]。本研究收集的家系有2例患者,均由MFN2基因突变所致,主要表现为缓慢进展的双下肢对称性肌肉无力伴跟腱反射消失、足部畸形。本家系所涉及患者均有弓形足,形成原因不清,有学者推测可能与疾病早期内侧与小腿的肌肉不平衡萎缩有关[14]。家系谱特点为连续两代出现同样疾病受累,性别无明显差异,此家系符合常染色体显性遗传模式,外显率为100%。
本例家系先证者神经电生理检查显示感觉及运动神经均出现了传导速度减慢,波幅降低,以CMAP波幅降低为著,多条神经不能引出波形,为脱髓鞘和轴索混合性改变。临床上通常将神经传导速度作为鉴别腓骨肌萎缩症脱髓鞘型(CMT1)与轴索型(CMT2)的重要依据之一。本研究中先证者出现CMAP波幅下降伴神经传导速度明显减慢。由于CMT是一种遗传性慢性进行性疾病,随着病程延长、周围神经损害加重,无论是周围神经脱髓鞘还是轴索变性,均可能出现神经传导速度显著减慢伴CMAP波幅下降甚至不能引出波形,故此时仅依据神经电生理参数难以鉴别原发病变是脱髓鞘性还是轴索性损害。因此,明确CMT的分型仍需依靠基因诊断。
CMT2型主要遗传方式为常染色体显性遗传,占CMT的20%~40%左右,其中最常见类型为MFN2基因突变导致的CMT2A2A型,约占8%~33.3%。MFN2基因定位于人类染色体1p36.2区,全长33kb,包含17个外显子,由757个氨基酸残基组成,位于线粒体外膜上并且两次跨线粒体外膜。编码的线粒体融合蛋白2广泛表达于心肌、骨骼肌等处,此蛋白可调节线粒体的融合,维持其融合与裂解动态平衡,在线粒体代谢过程中维持其网状结构。最常见致病机理为点突变。MFN2基因突变引起的过度表达或过少表达均可导致线粒体能量代谢的动态平衡紊乱,从而影响神经元营养物质和结构蛋白向轴突的运输,导致轴突发生变性而引起CMT2A2A。在本研究中,我们发现患病家系中所有患者的MFN2基因均发生第11外显子c.1066A>G的杂合错义突变。经检索c.1066A>G突变为已报道的致病性突变。p.T356A突变利用生物信息分析软件PolyPhen-2和MutationTaster预测蛋白功能及物种保守性,其预测结果为致病性,物种保守性分析显示,MFN2蛋白第356位氨基酸在不同的物种间几乎完全相同,表明在进化上高度保守。
综上所述,本家系具有显性遗传2A2A型CMT的临床特点,MFN2基因c.1066A>G(p.T356A)突变为其致病性突变。目前尚无有效的治疗手段,主要是对症和支持疗法,可采用理疗、按摩及肢体功能训练等康复医疗措施。由于本病病情进展缓慢,预后相对良好,及时诊断和治疗可延缓疾病的发展,注意避免使用对神经有害的药物以提高患者的生活品质。另外,认识该病特点、神经电生理表现和基因检测有助于早期确诊、早期治疗,为遗传咨询提供依据。
