分子尺度反应动力学模型构建在催化裂化/裂解过程中的应用进展
2021-04-20刘东阳白宇恩张霖宙张宇豪赵亮高金森徐春明
刘东阳,白宇恩,张霖宙,张宇豪,赵亮,高金森,徐春明
(中国石油大学(北京)重质油国家重点实验室,北京102249)
催化裂化反应是在热和催化剂的作用下使重质油发生裂化反应生成裂化气、汽油和柴油,催化裂解反应主要是在催化剂存在的条件下将轻质油进行高温裂解转变为低碳烯烃(乙烯、丙烯和丁烯)。目前,国内外科研机构和单位针对催化裂化/裂解反应的原料组成、反应器类型和产品分布的特点开发了一系列成熟的催化裂化/裂解工艺过程[1-3]。据统计,我国催化裂化所产汽油占汽油调和池的70%左右,所产柴油约占柴油调和总量的40%,催化裂化/裂解过程所产丙烯约占丙烯总产能的33%[4-5]。由此可见,催化裂化/裂解过程对我国交通运输业和化学工业的发展具有至关重要的作用。近年来,随着节能环保法规的日益严格、燃料油品质的快速升级以及低碳烯烃需求的不断增加,致使催化裂化/裂解过程产品分布和性质预测模型的开发变得尤为重要。催化裂化/裂解过程模拟的主要挑战集中在不同操作条件下将原料组成与产品收率和性质进行关联,分子尺度反应动力学模型是实现这一目标的有效途径,其本质是通过分子层次管控使石油馏分中每个分子的价值最大化[6-7]。
与经验模型和集总动力学模型相比,分子尺度反应动力学模型的研究细化到了石油烃类分子层面,是在对反应机理和反应类型研究的基础上,基于每种分子及其反应构建的反应动力学模型[8-9]。催化裂化/裂解过程分子尺度反应动力学模型可以对原料性质、催化剂、操作条件和产品组成相关联,对产品分布及性质作出精准预测,可实现对催化裂化/裂解工艺过程催化剂的选用和改进、工艺条件的优化、流程模拟和工艺工程放大[10-11]。
本文综述了催化裂化/裂解反应机理、反应类型、分子尺度动力学模型构建方法及其在催化裂化/裂解过程中的应用进展,具体包括结构化模型(structural model)[12-13]、单元步骤模型(single-event)[14]、键电矩阵模型(bond-electron matrix)[15-16]、结构导向集总模型(structure-oriented lumping)[17]和结构单元-键电矩阵混合框架(hybrid structural unit and bond-electron matrix)[18],并针对不同模型构建方法的优缺点进行了分析,期望对今后的催化裂化/裂解过程分子尺度反应动力学模型研究提供参考。
1 催化裂化/裂解反应机理和反应类型
分子尺度反应动力学模型构建需要通过反应规则描述复杂反应体系中每个分子的反应规律,而反应规则是在反应机理和反应类型的基础上建立起来的。另外,不同碳数、类型的石油烃类分子在催化裂化/裂解过程具有不同的反应规则,反应规则的制定与反应网络和反应动力学模型密切相关。因此,针对复杂多样的催化裂化/裂解过程原料组成,对催化裂化/裂解反应机理和发生反应类型的正确认识并建立准确的反应规则是构建分子尺度反应动力学模型的前提。
1.1 反应机理
石油烃催化裂化/裂解过程同时发生酸催化裂化和热裂化反应,两个反应分别遵循正碳离子和自由基机理。一般而言,在酸性分子筛催化剂上低温反应的情况下,反应过程遵循正碳离子机理,催化裂化/裂解反应发生在分子筛的Brønsted 酸中心[19]。碳正离子是催化裂化/裂解反应中形成的不稳定中间体,包括经典三配位碳正离子和非经典五配位的碳正离子[20-22]。其中,三配位碳正离子一般由烯烃分子经质子化反应生成,而五配位碳正离子是由氢质子直接攻击烷烃形成[23-24]。当反应温度超过650℃及金属氧化物存在条件下,石油烃类裂解过程发生自由基机理[25],如图1所示,经过自由基链引发、链传递和链终止三个阶段,最终形成稳定的产物分子[26]。

图1 烃类热裂解反应机理[26]
1.2 反应类型
石油烃分子催化裂化/裂解反应遵循平行-顺序反应原则,如图2所示[27]。

图2 催化裂化过程平行-顺序反应
当原料发生催化裂化/裂解反应时,朝着几个方向进行,同时生成中间馏分油、汽油和裂化气,这种反应称为平行反应;随着反应深度的增加,中间产物会继续反应,如中间馏分油和汽油会进一步裂解成裂化气,发生顺序反应。平行-顺序反应的一个重要特点,是反应深度对产品分布和收率有着重要的影响,随反应时间增加,裂解深度增加,最终产物裂化气和焦炭的含量会一直增加;而对于中间产物,到一定的反应深度后其裂化速率大于其生成速率,表现为其产率在开始一段时间内增大,但经过一最高点后开始下降。
1.2.1 催化裂化/裂解过程中的总包反应
表1 列出了不同族组成烃类分子的催化裂化/裂解总包反应,这些反应构成了复杂的催化裂化/裂解反应体系,不同烃类型裂解的难易程度顺序为:烯烃>正构烷烃>异构烷烃>环烷烃>芳烃。此外,随着原料组成碳链的增长,其键能下降,裂解反应越容易发生[28]。
1.2.2 催化裂化/裂解过程中的基本步骤
催化裂化/裂解反应中的基本步骤主要包括(去)质子化、β-裂化、烷基化、异构化、负氢离子转移和质子化裂化等[29],下面对石油烃类分子催化裂化/裂解反应中的基本步骤进行分类介绍。

表1 典型的催化裂化/裂解反应
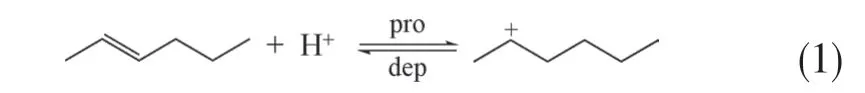
(1)质子化和去质子化反应 质子化(pro)和去质子化(dep)互为逆反应,质子化过程是烯烃与分子筛上的Brønsted酸中心相互作用形成碳正离子的过程;去质子化反应为碳正离子在分子筛上脱附生成烯烃的过程。式(1)表示C6烯烃的(去)质子化过程。
(2)β裂化和烷基化反应 在碳正离子的β位发生断裂生成新的更小的碳正离子和烯烃为β-裂化反应,新形成的碳正离子会继续发生β-裂化反应。烷基化(alk)反应为较小的碳正离子与烯烃分子相互作用形成较大的碳正离子的过程,烷基化反应与β-裂化反应互为一对逆反应,反应过程如式(2)所示。

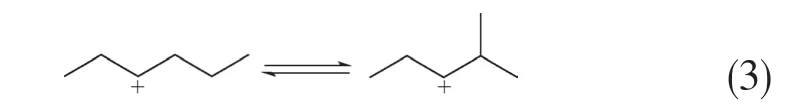
(3)PCP异构化反应 PCP异构化反应在催化裂化/裂解过程中广泛存在,可以提高产品的支化度,产物中的异构烷烃和异构烯烃主要是由该反应生成的,反应过程如式(3)所示。
(4)负氢离子转移反应 式(4)为负氢离子转移过程,烷烃中的负氢离子转移到正碳离子上,同时烷烃形成正碳离子,该反应是链传播的主要反应。

(5)质子化裂化反应 以异己烷为例的质子化裂化(prsc)过程如式(5)所示,异己烷受到来自分子筛Brønsted酸中心的氢质子攻击形成碳正离子和烷烃分子,随后,碳正离子从分子筛上脱附形成烯烃。

催化裂化和催化裂解反应机理基本一致,但催化裂解反应温度比催化裂化要高出几十摄氏度,且两者采用催化剂特点不同而导致反应速率存在差异。对于分子尺度动力学模型的构建,全反应网络的获取是基础,根据上述催化裂化/裂解反应机理和发生的反应类型制定反应规则,作为获取全反应网络的准则,最后对动力学参数调校得到分子尺度反应动力学模型,实现对产品分子组成的精准预测。
2 分子尺度反应动力学模型构建及其在催化裂化/裂解过程中的应用
分子尺度反应动力学模型的提出已有近三十年时间,不同国家和机构的学者开发了不同的模型,包括结构化模型、单元步骤模型、键电矩阵模型、结构导向集总模型和结构单元耦合原子拓扑矩阵模型。研究人员将上述模型应用到催化裂化/裂解反应体系中,目前已取得较好的模型预测效果。
2.1 结构化模型
1989 年,美国加州大学的Liguras 和Allen[12-13]提出了一种以虚拟分子代替复杂原料油组成的结构化模型,他们把石油中每个分子均看作是由几种碳中心的组合,通过基团贡献法预测产品性质。例如,用CH3、CH2两类碳中心表示正构烷烃,CH3、CH2和CH 三类碳中心表示异构烷烃,每一个碳中心的反应速率是碳数和碳中心所在位置的函数。据此可以描述原料中每一个分子,而通过纯化合物的催化裂化反应数据可以推算出不同碳中心的反应特性。通过上述方法对5 种不同的烃类,即正构烷烃、异构烷烃、烯烃、环烷烃和芳烃的反应特性进行研究,生成反应网络,推算出各类反应的速率常数。这样就将复杂反应体系构建成基于石油烃类分子碳中心和碳数的反应动力学模型。反应速率方程通过式(6)、式(7)求解。
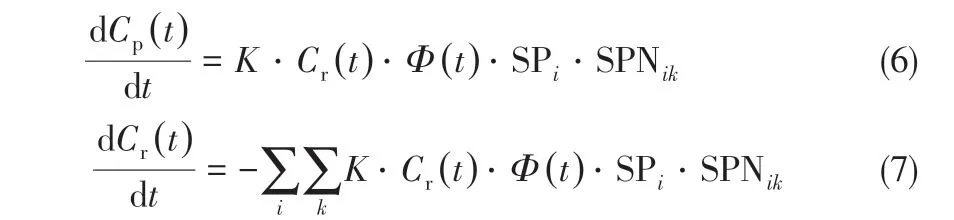
式中,Cr(t)为反应物浓度;Cp(t)为产物浓度;K为总反应速率常数;Ф(t)为催化剂时变失活函数;SPi为在反应物的第i个碳上形成碳正离子的概率;SPNik为反应物的k键断裂,在第i个碳上形成碳正离子的概率。
Liguras和Allen等[12-3]依据结构化模型方法,选取了正十六烷、2,7 二甲基辛烷和异丙基环己烷进行裂化反应获取实验数据,推算出不同碳数、不同类型的碳中心的反应速率常数。并对正十六烷裂解产物分布进行预测,预测结果与实验数据如表2所示。研究还表明模型对碳数分布较为敏感,而对碳中心分布敏感性差。这表明对原料分析表征时,按碳数分布的气相色谱-质谱分析比核磁分析更有利。

表2 正十六烷裂解产物组成(物质的量分数)
2.2 单元步骤模型
20世纪80年代末,比利时根特大学的Froment等[14,30]提出了采用单元步骤法对复杂反应体系构建动力学模型,即单元步骤动力学模型。单元步骤动力学模型构建结合正碳离子理论和过渡状态理论,将复杂的酸催化反应按基本步骤进行简化,分为质子化、去质子化、烷基化、β-裂化、负氢离子转移等等。单元步骤模型采用布尔邻接矩阵和辅助向量表示石油烃类分子和碳正离子,见图3,图中1表示相邻两个碳原子间存在C—C 化学键,0 表示没有C—C 化学键,完整反应网络由布尔关系矩阵算法生成。图3表示2-甲基丙烯基环己烷的布尔邻接矩阵,图中第1个辅助向量“=”表示双键位置,第2 个辅助向量“+”表示正电荷位置[31]。Froment等利用过渡态理论,将分子的结构对反应活化熵变的贡献从反应速率常数中提取出来,命名为单事件数,剩余的部分(单事件速率常数)则假设仅与发生基本步骤类型有关。反应速率常数如式(8)所示。通过求得的速率常数进而预测产品分布及性质。

式中,ne称为单事件数;称为单事件速率常数。
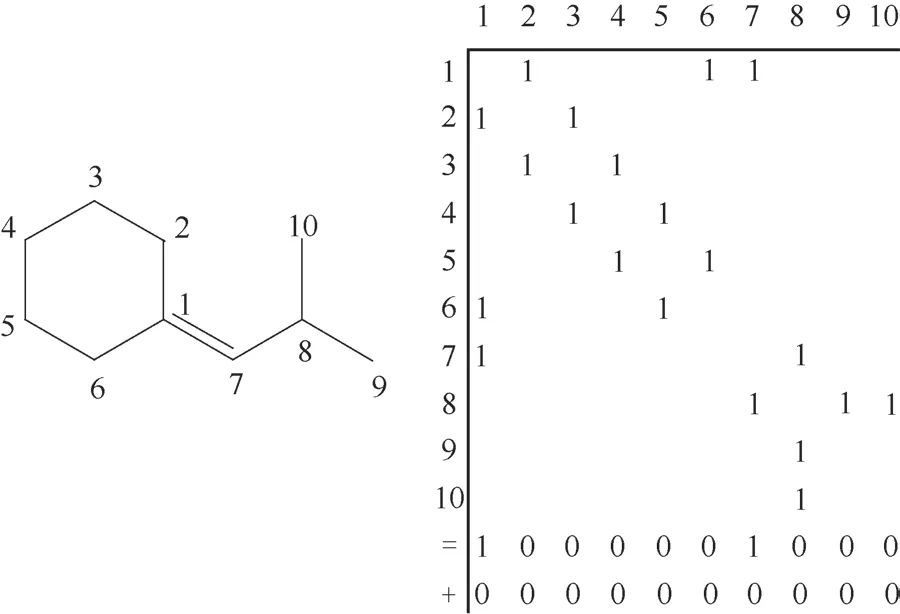
图3 2-甲基丙烯基环己烷布尔邻接矩阵[31]
Feng 等[31]于1993 年首次将单元步骤法应用催化裂化反应体系的分子动力学模型构建,建立了正庚烷催化裂化的分子尺度反应动力学模型,利用计算机算法生成了基于反应中每一个反应物分子和碳正离子的反应网络,通过正庚烷裂解数据求取了单事件速率常数。随后,Dewachtere 等[32]将Feng等建立的动力学模型耦合到提升管反应器模型中,并重点研究了进料喷嘴对产品中干气、焦炭收率和提升管反应器轴向温度的影响,实现了催化裂化分子反应动力学耦合提升管反应器的模型开发。Quintana-Solórzano 等[33]利用正癸烷、甲基环己烷、丁基环己烷和辛烯作为混合原料进行催化裂解实验,采用单元步骤法,通过裂解实验数据对动力学参数进行拟合,成功构建了单元步骤动力学模型,并实现对不同温度下的产物分布进行有效预测。
Marin 等[34]采用单元步骤模型,建立了一种焦炭形成的动力学模型,该模型计算出的产品和焦炭收率曲线具有典型的工业应用价值。随后,Marin课题组[35]对693~753K 下(环)烷烃/1-辛烯混合物催化裂化的初始结焦速率进行了模型构建,引入了烃转化的动力学速率方程和焦炭对催化剂活性和选择性影响的全局速率方程的组合,实现了催化裂化中的裂化反应和焦炭形成过程模型的同时建立。Van Borm等[36]通过引入催化剂酸度的描述研究了同类型的不同酸性的催化剂对烷烃催化裂化反应体系的影响,该模型能很好地描述实验观察到的趋势。
2.3 键电矩阵
特拉华大学的Klein 课题组[37]采用键电矩阵的方法形成了分子管理的软件平台:Kinetic Modeler's Toolbox(KMT)。该方法是根据化学反应是旧键断裂和新键生成的原则,提出以矩阵表示复杂的石油分子,以“+”表示化学键的生成,“-”表示化学键的断裂。采用该模型,可以在机理层面建立反应动力学模型,图4展示了基于键电矩阵方法乙烷裂解的化学反应自动生成方法[38]。

图4 乙烷裂解键电矩阵表示[38]
Watson 等[39-41]以正辛基环己烷和正庚烷等作为模型化合物,采用键电矩阵的方法建立了催化裂化过程机理层面的反应动力学模型,该模型基于反应族和线性自由能方程(LFERs)对反应速率常数进行评估。一个化合物裂解的基本步骤仅由纯组分实验确定的定量结构-反应关系(QSRCs)的限制,并通过计算机编程对模型化合物的裂解实验数据拟合,求取模型中的参数,最终完成了基于键电矩阵方法的分子尺度反应动力学模型构建。通过上述方法可使模型方法降到可控范围,同时满足模型求解的可行性与拟合结果的准确性。
2.4 结构导向集总
Mobil 公司的Jaffe 和Quann 于1992 年提出结构导向集总(SOL)方法[17]。该方法采用结构向量代表复杂的石油烃分子,他们提出22 个特征结构单元,如表3所示。通过改变这些结构单元的排列组合可以很好地构造出各种石油烃类分子。
Christensen 等[42]采用SOL 方法建立了固定床催化裂化工艺模型,该模型用了3000 种分子来描述原料组成,并且描述了60 种反应规则。它根据反应速度常数和催化剂活性衰减速度常数来表征催化剂的裂解行为,成功计算了不同原料和不同催化剂下的产物收率。
李春义等[43]采用SOL方法表示反应物和生成物的结构,将SOL 与蒙特卡罗方法相结合,构建了FCC 汽油催化裂解过程的分子尺度反应动力学模型。该模型用2000 个分子表示FCC 汽油的原料组成,根据模型化合物的催化裂解行为求取模型所需的反应速率常数。该模型的预测值与真实值相对误差小于10%。Yang 等[44]采用SOL 和蒙特卡罗方法,通过60 项反应规则建立了催化裂化汽油二次裂化反应网络,并采用蒙特卡罗方法计算了各烃类分子的反应概率,得到产物的组成分布。之后,以我国工业催化裂化装置的3个催化裂化汽油样品作为模拟原料,验证了该方法的有效性。此外,华东理工大学采用结构导向集总方法对不同原料和不同工艺的催化裂化/裂解过程进行了模型构建。翁惠新等[45-46]采用SOL 和蒙特卡罗相结合的方法对DCC(Deep Catalytic Cracking)工艺进行模型构建。其中,蒙特卡罗模拟方法模拟了虚拟分子组成和性质,建立的动力学模型预测值与试验值最大相对偏差<10%。陈华和刘纪昌等[47-48]根据催化裂化反应机理和MIP(Maximizing Iso-Paraffin)工艺过程特点,构建了基于SOL 方法分子尺度反应动力学模型,模型预测值与工业数据误差为1.0%左右。之后,该研究团队将该模型用于中国石化某分公司的MIP 装置的工艺操作条件优化。Zhu 等[49]采用SOL方法建立了减压瓦斯油FCC 分子尺度反应动力学模型,他们通过最小二乘法计算了产物的含量及分布。该模型预测的产品分布、性质与实验数据吻合较好。最近,清华大学邱彤课题组[50]提出了SOL和基团贡献法的分子尺度反应动力学模型。以SOL表示石油烃类分子,基团贡献法用于评价其性质,并且提出了三步重建算法,包括结构分布参数优化、利用优化后的参数建立分子文库和利用最大信息熵方法得到分子组分。
2.5 结构单元-键电矩阵
鉴于ExxonMobil公司的结构导向集总方法和特拉华大学Klein 课题组提出的键电矩阵方法,中国石油大学(北京)分子管理课题组[8,51]提出结构单元和键电矩阵混合的方法(SU-BEM),开发了分子管理平台。该方法底层为键电矩阵,表层为结构单元,为了更适合复杂的石油分子,研究团队将结构单元分为核心结构单元和侧链结构单元,修改并增加了部分结构单元,图5为SU-BEM框架的34个结构单元(CUP结构单元)[52]。
反应网络的生成是分子尺度反应动力学模型构建的基础,SU-BEM框架采用确定性网络生成算法得到反应网络[53],即反应物分子和新生成的产物分子遍历每个反应规则,重复迭代直至没有新产物生成,便得到全反应网络。该模型选择阿伦尼乌斯方程 或 LHHW (Langmuir Hinshelwood Hougen Watson)方程形式求取反应速率常数,如式(9)和式(10)所示。化学反应转化为动力学常微分方程组(ODEs)的流程如图6所示[52]。


图5 SU-BEM框架的结构单元

图6 反应网络与常微分方程组转化[52]
Chen等[18]通过SU-BEM方法构建了耦合提升管反应器模型的催化裂化分子尺度动力学模型。利用线性自由能关系(LFER)和定量结构活性关联(QSRC)减少模型参数,采用工业数据对模型参数进行调整优化。反应器模型包含反应器类型、温度分布、相行为、质量平衡等方程。通过上述模型研究了各馏分在反应器轴向分布情况以及反应温度对各馏分收率的影响,并对汽油馏分的关键性质进行预测,该模型实现了超过3800 个分子及7500 个化学反应的复杂反应网络构建。模型计算值与试验值之间有很好的一致性,为催化裂化过程的优化提供了更加精细的技术方案。
3 分析与讨论
催化裂化反应体系分子尺度反应动力学模型构建首次是通过结构化模型实现的,模型将石油烃类分子简化为几种碳中心的组合,实现了通过相对较少的虚拟分子表示复杂的原料组成,降低了复杂原料油动力学模型构建的难度。根据正碳离子反应机理,利用单体烃的裂化数据求取每一个碳中心的反应速率常数,打破了集总动力学模型的传统思路。然而,该模型以反应物和产物分子间的总包反应为研究对象,模型中仍保留了一定程度的集总。另外,仅通过CH3、CH2、CH 代替复杂的催化裂化/裂解过程原料组成,可靠性较差,仍需进一步改进[54]。结构化模型是在原料分析的基础上建立起来的,对于仪器难于表征的复杂重质原料油很难实现模型构建。因此,该模型更适用于仪器分析可知的轻质原料。虽然结构化模型比集总动力学模型能够预测和描述更多的产物信息,但它们仍使用传统意义上的集总概念,致使模型参数仍与进料组成和性质有一定的关系,这就大大限制了模型的外推性。
与结构化模型不同的是,单元步骤模型可以将复杂的酸催化反应归纳为几种基本步骤,是机理层面的反应动力学模型,且与原料的组成和性质无关[29]。另外,单元步骤模型适用于所有的复杂酸催化反应,在石油炼制与化工领域有着广泛的应用前景。但对于重质原油的模型构建,由于体系过于复杂,很难从机理层面来对反应过程进行完整的模型建立。所以对于较重的原料加工过程中,单元步骤模型构建方法会受到一定程度的限制。
从目前的研究情况来看,键电矩阵法在催化裂化/裂解过程中的应用较少,该方法主要优点在于能对机理层面和路径层面的反应过程建模,可根据使用者的需求,构建不同详细程度的模型。但缺点是,对于采用矩阵表示复杂石油烃类分子使建模过程和反应网络变得复杂,导致键电矩阵法在重质原料油中的推广和应用受限[55]。
Mobil 公司的SOL 模型已经成功应用于工业生产,但该模型的开发需要大量的实验和工业数据做支撑。结构导向集总动力学模型构建过程比键电矩阵法更简单,与结构化模型相比,可以细致地表达出反应中的每一个分子的结构特征。此外,SOL中的结构单元具有扩展性,研究者可以直观的对结构向量进行修改和删减以适用不同的原料组成。在模型精确度方面,由于SOL无法区分同分异构体,导致轻质油品建立模型的精确度没有键电矩阵法高。相比其他建模方法,对于重质原料油的SOL模型构建方法更简单,但该方法只能做到路径层面而无法深入到机理层面,导致模型的预测能力比机理层面的模型精度差。而且SOL模型还保留了一定程度的集总,导致模型的参数仍与进料组成有关。
中国石油大学(北京)开发的分子管理平台底层建立在键电矩阵的基础上,在设计结构单元之后,将其与键电矩阵进行映射。SU-BEM可实现反应网络的自动生成及可视化,并通过计算机实现了化学反应网络与反应速率表达式的切换。该反应动力学建模方法可实现机理层面和路径层面的双重建模,适用于分子管理不同层次的需求(用户友好型和计算机编程水平),具有较强的使用性。
分子尺度动力学模型在分子层次上将原料与产物的组成和性质相关联,使人们对化学反应的认识从馏分-结构族层次上升到分子级别。相比传统以集总划分的动力学模型,分子尺度反应动力学模型可以实现石油分子的高效管理和开发利用。上述几种动力学模型基本使用矩阵或结构单元等图论形式表示石油分子,以方便计算机读取和计算。其中,结构化模型、单元步骤模型、键电矩阵法更适用于轻、中馏分油的模型构建;SOL、SU-BEM 则能够通用于轻、中、重的石油加工过程中。
关于分子尺度动力学模型反应速率常数的求解均依赖于计算机,目前已经形成很多动力学模型求解的软件包,包括CVODE、DASSL,其中CVODE由C 语言编写,提供C、C++、Fortran、Matlab 等接口,DASSL由Fortran 编写[56]。图7展示了各层次模型的预测能力与复杂度(计算耗时)间的关系[52]。过于详细的动力学模型虽然能更加接近真实的反应过程,但是过长的求解时间将导致无法对模型进行精确的优化。而过于简单的动力学模型则无法反映分子的真实转化行为,脱离了分子尺度反应动力学模型的初衷。因此,对于轻质油品来说,机理层面的模型对产品的分布和性质预测更为精准。对重油反应体系来讲,需要建立在保证计算效率前提下尽可能详细的分子尺度反应动力学模型,其中路径层面的模型较为适合。
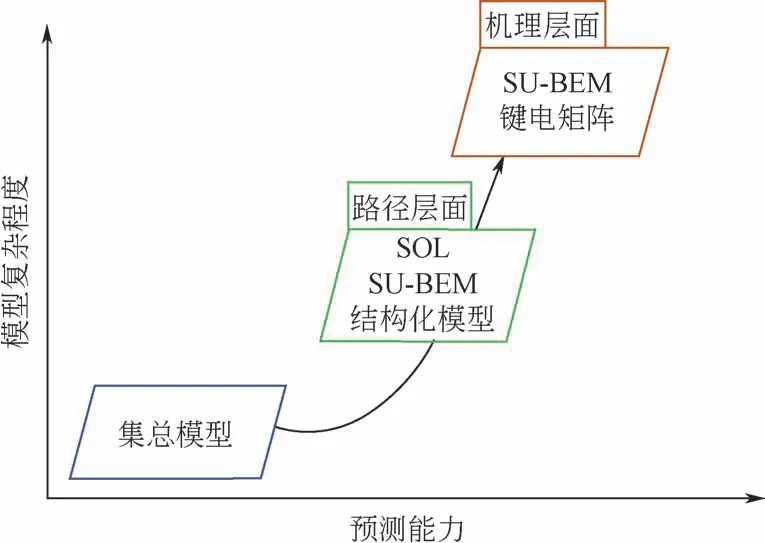
图7 反应动力学模型中模型复杂程度与预测能力的关系[52]
虽然分子尺度反应动力学模型拥有能够预测产物大量宏观性质的优势,但对重质原料油,仪器分析表征方法和计算效率一直制约着模型的发展和应用。对于现有分析技术无法表征的石油烃分子,一般用蒙特卡罗(Monte Carlo)随机抽样分子集构建和最大信息熵法。最大信息熵法计算迅速,适合对原料体系有一定认识的情况[57-61]。后续应继续开发石油分子分析表征技术,对原料的认识更精准,从而构建更加高效、准确的反应网络和动力学模型。目前,关于分子尺度反应动力学研究的软件有KMT[37]、RMG[62]、Gensys[63]、RING[64-67]等,发 展 比较完善的是KMT 软件,涵盖了全反应网络的构建和ODEs方程的求解等模块。
4 结语与展望
催化裂化/裂解过程是石油炼制与化工行业重要的催化反应工艺,分子尺度反应动力学模型对该复杂反应体系产品预测、催化剂选用与改进、工艺工程放大具有重要的指导意义,可以反映催化裂化/裂解过程的分子层次信息,实现对产品质量及加工链条的精确管理。现有分子尺度动力学模型的开发均采用图论的形式表示石油分子,对于速率方程的求解均借助计算工具进行语言编程。
催化裂化/裂解过程分子尺度反应动力学模型虽然能更为精确的描述化学反应本质信息,对产品分布及性质进行较为精准的预测。但对于高碳数、重质石油烃类的原料,由于现有分析条件不足,难以得到详细的分子组成,需用虚拟的分子性质代替原料组成,虚拟分子还存在着一定程度上的集总,导致模型构建会有偏差。此外,重质油品组分数较多,使计算机解常微分方程所耗费的机时过大。因此,随着计算机和分析表征技术的发展,开发更为细致的分子尺度反应动力学模型是该领域发展的趋势。目前,催化裂化/裂解过程工业装置大部分为提升管或固定床反应器,催化裂化/裂解过程催化剂的结焦直接影响催化剂的活性和产物分布,催化剂结焦速率和产品分布、性质以及催化剂性能密切相关。由此可知,建立与催化剂失活和反应器数学模型相耦合的反应动力学模型可以更好的和复杂催化裂化/裂解反应体系相吻合。
分子尺度反应动力学模型是石油炼化分子管理的重要组成部分,也是石油炼制与化工行业向智能化、高效化和集成化发展的方向。动力学模型的构建最终还是立足于炼厂的实际应用,机理层面的模型比路径层面的模型更为精确,但模型复杂程度大,计算耗时时间长。因此,选用什么类型的动力学模型要根据实际的情况而定。此外,建立对分子集构建,反应网络构建和动力学参数求解的集成化平台是分子尺度动力学发展的必然趋势。
