三氯乙酰亚胺酯法合成紫草素-1′-O-β-D-吡喃葡萄糖苷
2021-04-20李龙珠张玉心吴成柱李红梅
李龙珠,张玉心,吴成柱,李红梅,马 涛
紫草素是从我国传统中药紫草中分离得到的一种脂溶性萘醌类化合物[1],是紫草的主要活性成分。研究表明紫草素具有抗肿瘤[2-4]、抗炎[5]、抗病毒[6]、抗菌[7]、促进伤口愈合[8]等药理作用。与其他萘醌类化合物类似,紫草素的稳定性较差,且在水中的溶解度低,这在一定程度上阻碍了紫草素的开发与应用[9]。
糖是自然界中存在最为广泛的生物分子,几乎存在于所有生命体中,通过糖苷化修饰可以提高化合物的水溶性和稳定性[10-11]。本课题组前期采用酶催化法已经制得两种紫草素吡喃葡萄糖苷,其稳定性均显著高于紫草素,水溶性相较紫草素增加上千倍,两种紫草素吡喃葡萄糖苷在体外抗肿瘤作用相较紫草素有所下降[12],但在后续开展的肝癌裸鼠移植瘤预实验中,紫草素-1′-O-β-D-吡喃葡萄糖苷的抑肿瘤作用明显优于紫草素。为了对其体内抗肿瘤作用进行全面研究,需要大量制备紫草素-1′-O-β-D-吡喃葡萄糖苷用于裸鼠移植瘤实验及药代动力学研究。但是在实验室条件下,通过酶催化法所得糖苷化合物的量只有微克级,难以满足动物实验的需要,而且酶的制备需要经过菌群培养、基因表达、构建重组质粒、提纯等多个步骤,这也导致了酶的制备成本高[13],因此迫切需要建立一种成本低、操作简便且适于大量制备紫草素-1′-O-β-D-吡喃葡萄糖苷的方法。
目前已有研究[14]采用三氯乙酰亚胺酯法将多种乙酰基取代的糖基加在紫草素1′-OH上,制得了11种紫草素苷,尚未见化学法合成紫草素-1′-O-β-D-吡喃葡萄糖苷的报道。本研究选择β-D-吡喃葡萄糖为起始原料,经选择性乙酰化后,再进一步制得糖基三氯乙酰亚胺酯作为糖苷供体,选择催化效能更佳的三氟甲磺酸三甲基硅脂来催化紫草素糖基化反应,制得紫草素-2,3,4,6-四-O-乙酰基-β-D-吡喃葡萄糖苷,利用甲醇钠/甲醇醇解脱乙酰基后制得紫草素-1′-O-β-D-吡喃葡萄糖苷,并采用核磁共振法、质谱法对各产物的结构进行了鉴定。
1 材料与方法
1.1 仪器与试药 IKA RV10型旋转蒸发仪(德国 IKA 公司);LC-20A型高效液相色谱仪(日本岛津公司);B23-2型恒温磁力搅拌器(上海硕光司乐仪器有限公司);梅特勒-托利多XP250型天平(瑞士梅特勒-托利多公司);BRUKER ASCENDTM400 MHz核磁共振光谱仪(美国Bruker公司);Thermo TSQ quantis液质联用仪(美国Thermo Fisher Scientific公司)。
甲醇、乙酸乙酯、石油醚、乙醇、无水乙醚(上海泰坦化学有限公司)均为分析纯;左旋紫草素标准品(中国药品生物制品检定所,批号110769-200506);乙酸酐(江苏彤晟化学试剂有限公司);4A分子筛、吡啶(萨恩化学技术有限公司);β-D-葡萄糖、四氢呋喃、1,8-二氮双环(5,4,0)-7-十一烯、三氯乙腈、乙酸肼、碳酸氢钠(上海麦克林生化科技有限公司);三氟甲磺酸三甲基硅脂(阿达玛斯试剂有限公司)。
1.2 方法 以二氯甲烷为溶剂,三氟甲磺酸三甲基硅脂为催化剂,在严格无水和N2保护下,通过自制的2,3,4,6-四-O-乙酰基-β-D吡喃糖酰基2,2,2-三氯乙酰亚胺酯与紫草素发生糖苷化反应,有效合成了紫草素-2,3,4,6-四-O-乙酰基-β-D-吡喃葡萄糖苷,并在甲醇钠/甲醇体系中将乙酰基醇解为羟基,最终得到目的产物紫草素-1′-O-β-D-吡喃葡萄糖苷(见图1)。
1.2.1 1,2,3,4,6-五-O-乙酰基葡萄糖(1)的合成 取乙酸酐56.7 mL和吡啶5 mL混匀后分多次加入至18 gβ-D-葡萄糖中,冰浴条件下搅拌,并缓慢升至室温反应过夜。反应结束加入25 mL蒸馏水搅拌,再用25 mL乙酸乙酯萃取2~3次,取有机相用10%饱和碳酸氢钠水溶液中和反应产生的副产物醋酸及过量的乙酸酐,再用水洗3次,每次20 mL,上层有机层用旋转蒸发仪除去溶剂,得到的粗产物于室温下用75 mL乙醇结晶,得到白色粉末,采用核磁共振波谱(nuclear magnetic resonance spectroscopy,NMR)和电喷雾-质谱(electrospray ionization mass spectrometry,ESI-MS)表征其结构。
1.2.2 2,3,4,6-四-O-乙酰基-β-D-吡喃葡萄糖(2)的合成 将20 g化合物(1)和5.2 g乙酸肼加入到10 mL四氢呋喃中溶解,室温下反应4 h,旋转蒸发除去溶剂,用10 mL二氯甲烷溶解残余物,将有机相用水洗3次,每次5 mL,旋转蒸发除去溶剂得粗产物,加入20 mL无水乙醚和20 mL石油醚后于冰水浴下析出结晶,得到白色粉末,采用NMR和ESI-MS表征其结构。
1.2.3 2,3,4,6-四-O-乙酰基-β-D吡喃糖酰基2,2,2-三氯乙酰亚胺酯(3)的合成 取9 g化合物(2)和10 mL三氯乙腈依次加入至有50 mL无水二氯甲烷的烧瓶中,冰浴条件下搅拌30 min,再缓慢滴加1.5 mL的1,8-二氮双环(5,4,0)-7-十一烯,撤除冰浴,在N2保护下室温反应过夜。旋转蒸发溶剂后得黄褐色油状物,油状物经硅胶柱层析(石油醚∶乙酸乙酯=8∶1)纯化,最终得白色粉末,采用NMR和ESI-MS表征其结构。
1.2.4 紫草素-2,3,4,6-四-O-乙酰基-β-D-吡喃葡萄糖苷(4)的合成 将10 g紫草素和18.8 g化合物(3)溶解于70 mL无水二氯甲烷中,加入4A分子筛粉末,N2保护下室温搅拌30 min,然后缓慢滴加0.63 mL三氟甲磺酸三甲基硅脂,于-20 ℃反应8 h。抽滤除去分子筛,将有机相用水洗3次,每次50 mL,旋转蒸发除去溶剂后,将粗产物用乙酸乙酯复溶,经硅胶柱层析(石油醚∶乙酸乙酯=5∶1)纯化,最终得红色粉末,采用NMR和ESI-MS表征其结构。
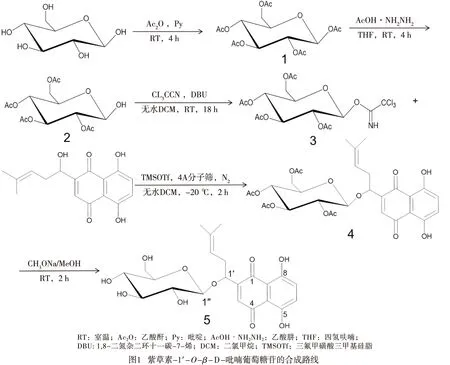
1.2.5 紫草素-1′-O-β-D-吡喃葡萄糖苷(5)的合成 取10 g化合物(4)溶解于50 mL甲醇中,向其中缓慢滴加甲醇钠溶液,调节溶液pH至8~9,室温回流反应2 h,加入冰醋酸调pH为中性并终止反应。旋转蒸发溶剂后,将粗产物用二氯甲烷复溶后经硅胶柱层析(二氯甲烷∶甲醇=10∶1)纯化,最终得红色粉末,采用NMR和ESI-MS表征其结构。
1.2.6 化合物5纯度测定 取适量化合物5,加入适量甲醇配制成溶液,取20 μL化合物5的甲醇溶液进高效液相色谱仪分析,以峰面积归一化法测定化合物5纯度。采用SunFireTMprep C18(10 mm×250 mm) 色谱柱,以水-乙腈作为流动相进行梯度洗脱(0~20 min,20%乙腈→100%乙腈),流速为1 mL/min,检测波长为254 nm。
2 结果
化合物1为白色粉末,得到28.5 g,收率73.1%。1H NMR (400 MHz,CDCl3)δ:5.73 (d,J=8.0 Hz,1H),5.27 (t,J=9.4 Hz,1H),5.15 (m,2H),4.30 (dd,J=4.6,12.5 Hz,1H),4.13 (dd,J=2.2,12.5 Hz,1H),3.86 (m,1H),2.13 (s,3H),2.10 (s,3H),2.05 (s,6H),2.03 (s,3H)。13C NMR(100 MHz,CDCl3)δ:170.6,170.1,169.4,169.2,168.9,91.7,72.8,72.7,70.2,67.8,61.5,20.8,20.7,20.6,20.6,20.5。ESI-MS(m/z):413.15[M+Na]+。
化合物2为白色粉末,得到12.8 g,收率71.7%。1H NMR (400 MHz,CDCl3)δ:5.27 (t,J=9.5 Hz,1H),5.10 (t,J=9.9 Hz,1H),4.90 (dd,J=8.0,9.7 Hz,1H),4.76 (t,J=8.6 Hz,1H),4.25 (m,2H),3.77 (m,1H),3.66 (d,J=8.8,1H),2.11 (d,J=1.4,6H),2.04 (d,J=3.8,6H)。13C NMR(100 MHz,CDCl3)δ:170.9,170.7,170.1,169.6,169.5,95.6,73.3,72.2,68.5,62.0,20.7,20.7,20.6,20.6。ESI-MS(m/z):371.13[M+Na]+。
化合物3为白色粉末,得到12.4 g,收率57.8%。1H NMR (400 MHz,CDCl3)δ:8.69 (s,1H),6.63 (d,J=4.0 Hz,1H),5.58 (dd,J=1.2,3.1 Hz,1H),5.45 (dd,J=3.1,10.8,1H),5.39 (dd,J=3.5,10.8 Hz,1H),4.46 (t,J=6.6 Hz,1H),4.15(m,2H),2.19 (s,3H),2.05 (s,3H),2.03 (s,3H),2.04(d,J=2.1,6H)。13C NMR (100 MHz,CDCl3)δ:170.3,170.1,170.1,169.9,161.0,93.6,90.8,69.0,67.5,67.4,66.9,61.2,20.7,20.6,20.6,20.5。ESI-MS(m/z):514.08[M+Na]+。
化合物4为红色粉末,得到8.7 g,收率68.3%。1H NMR (400 MHz,CDCl3)δ:12.48(s,1H),12.42 (s,1H),7.14 (m,3H),5.15 (m,1H),5.03 (m,3H),4.89 (m,1H),4.58 (d,J=8.1 Hz,1H),4.01 (m,2H),3.57 (m,1H),2.4(m,1H),2.24 (m,1H),2.12 (s,3H),1.98 (s,3H),1.95 (s,3H),1.87(s,3H),1.60 (s,3H),1.47 (s,3H)。13C NMR (100 MHz,CDCl3)δ:181.2,178.6,170.2,170.0,169.1,168.5,165.1,164.8,149.2,135.0,134.1,13.7,131.5,119.2,112.1,111.6,101.7,75.5,73.2,72.0,71.4,69.1,61.9,33.5,25.8,20.8,20.6,20.4,20.3,17.8。ESI-MS(m/z):641.26[M+Na]+。
化合物5为红色粉末,得到4.9 g,收率67.3%。1H NMR (400 MHz,CDCl3)δ:7.28(s,3H),7.21(s,3H),5.37(t,J=14.8,1H),5.14(br s,1H),4.64(d,J=7.7,1H),3.23(m,1H),3.26(m,2H),3.27(m,2H),3.53(m,1H),3.72(m,1H),2.63(s,1H),2.62(m,1H),2.64(m,1H),2.53(m,1H),1.66(s,3H),1.52(s,3H)。13C NMR (100 MHz,CDCl3)δ:183.5,183.3,175.2,174.4,151.7,136.8,135.6,135.0,133.8,120.7,115.6,113.5,104.7,79.6,78.4,75.2,73.5,71.8,63.8,35.5,26.7,18.8。ESI-MS(m/z):473.19[M+Na]+。
根据ESI-MS和NMR结果分析其分子式为C22H26O10,其共振谱与紫草素相似,除了来自葡萄糖基部分的额外核磁信号,基于葡萄糖异头位质子的耦合常数(为7.7 Hz),包括从δH 4.64(d,J=7.7,1H,H-1″)到δC 75.2(C-1′)的异核多键关联表明葡糖基部分已经连接到紫草素C-1′处,化合物5的结构被鉴定为紫草素-1′-O-β-D-吡喃葡萄糖苷。总收率为38.9%,峰面积归一化法测定其纯度为96.7%,色谱图见图2。
3 讨论
相较于酶催化法,化学合成法制备糖苷类化合物具有原料易得,成本低的优势。常用的化学糖基化方法主要有Koenig-Knorr法[15]、三氯乙酰亚胺酸酯法[16]和相转移催化法[17]等。Koenig-Knorr法产率低、区域选择性差、反应后处理繁琐,并且使用价格昂贵且环境污染较大的汞、银盐作催化剂,近年来该方法的使用在逐渐减少。相转移催化法具有反应速度快,反应条件温和,后处理简单的特点,但是Koenig-Knorr法和相转移催化法在合成糖基供体时常以苯乙酰基或乙酰基保护的溴代糖作中间体,溴代糖对制备和贮藏的条件要求十分苛刻,且极易分解,会给下一步的糖基化反应带来很大影响。

三氯乙酰亚胺酯法制备糖苷类化合物不需要重金属催化剂,多采用糖基三氯乙酰亚胺酯作为糖苷供体,与其他糖基给体相比,糖基三氯乙酰亚胺酯更适合于氧苷类化合物的合成且不易形成原酸酯副产物,由于有邻基参与效应,糖基三氯乙酰亚胺酯法具有高立体选择性和高收率的优点[18],近年来在寡糖和糖苷的合成中得到了广泛的应用,逐渐成为首选方法[13]。
三氯乙酰亚胺酯法对底物的要求较高,反应体系要严格除水,因此在合成化合物4时采用加入分子筛的方法以除去反应体系中的水,经过预试验探索,最终选择了除水效果最好的4A分子筛。在前期实验摸索时,采用了文献[14]中的三氟化硼乙醚溶液作为糖基化反应催化剂,反应得到较多副产物,目标产物产率低,不易纯化,后将催化剂更换为三氟甲磺酸三甲基硅脂,化合物4的得率显著提高,收率达到57.8%,且副产物减少。考虑到糖苷键易断裂,在化合物4除去乙酰基制备化合物5时不可采用水解的方式,本研究选择无水甲醇为溶剂,采用甲醇钠/甲醇体系对化合物进行醇解,且反应需要在低温、无水的条件下进行,达到减少脱水物杂质,提高产物收率的目的。
三氯乙酰亚胺酯法用于紫草素-1′-O-β-D-吡喃葡萄糖苷合成,具有反应温和、操作简单、高立体选择性的优点,也为其他天然物质的糖基化修饰提供了参考。
