安徽省水稻纹枯病菌的致病性及遗传多样性
2021-04-18汪章勋张立新檀根甲
侯 冕,汪章勋,张立新,李 淼,檀根甲
(安徽农业大学 植物保护学院,安徽 合肥 230036)
近年来,纹枯病的危害逐年加重,在我国南方长江中下游尤为严重[1-2]。目前,该菌被划分为14个融合群,共18个融合亚群,水稻纹枯病作为世界范围内最具毁灭性的真菌病害之一,隶属其中的AG1-ⅠA融合亚群[3],纹枯病主要以存活在土壤中的菌核作为初侵染源,11年以上的菌核仍有12%的萌发率[4],为农业生产带来重大危害。沿淮的水稻种植在安徽省的粮食总产量中占据着重要的地位[5]。明确病原菌遗传信息有助于推演出其进化规律,为病害的预警及抗病育种提供支持。
近年来,各种分子标记技术在植物病原菌的遗传多样性研究中得到广泛应用[6],其中ISSR分子标记是一种简单重复序列区间扩增多态性分子标记,能更好地揭示病原菌基因组的多态性,具有重复性好、信息量大且多态性高等优点。Goswami等[7]对印度北部的水稻纹枯病菌进行了ISSR分析;黄雯雯[8]采用RAPD对安徽省7个市县32株菌株进行分析,表明安徽省水稻纹枯病菌的遗传结构丰富。但上述研究菌株量较少,涉及县市不全面,所使用菌株是十年前采集,现今安徽省水稻纹枯病菌可能已产生较大变化。
本研究对安徽省的15个水稻主产市县的94个水稻纹枯病菌菌株进行ISSR遗传标记、致病性进行分析,以期明确其种群分布情况及不同地理来源的菌株的亲缘关系,为安徽省水稻纹枯病绿色防控提供基础。
1 材料与方法
1.1 供试菌株
2018—2020年7—9月从安徽省15个市县稻田采集具有水稻纹枯病病症的样品,采用周而勋的水琼脂快速分离法[9],共获得94株水稻纹枯病菌菌株(采集地点分布及数量见表1),转于试管中保存于4℃冰箱备用。

表1 水稻纹枯病菌菌株采集分布
1.2 供试水稻品种
徽两优996。
1.3 DNA的提取
每个菌株挑取一个直径5mm的菌蝶分别置于装有100mL PDB的250mL锥形瓶中,25℃、160r/min摇培3d,过滤获取菌丝,采用CTAB法[10]抽提各菌株基因组的DNA,用微量紫外/可见分光光度计(ND-1000,NanoDrop)测定DNA的浓度与质量,-20℃冰箱保存备用。
1.4 菌丝融合群鉴定
采用Matsumoto[11]设计的引物(表2)鉴定其所属融合群。将提取的DNA用表2引物进行PCR扩增,PCR反应程序为94℃预变性5min;94℃变性1min;54℃退火2min;72℃延伸3min;30个循环;72℃延伸5min。PCR结束后取5微升PCR产物在含有溴化乙锭(EB)的1.0%琼脂糖凝胶上电泳15min,使用凝胶成像系统观察,根据谱带有无鉴定其菌丝融合群。

表2 直接检测和鉴定R. solani中AG 1-IA,AG 1-IB,AG 1-IC,AG 2-1和AG 2-2分离株的引物
1.5 ISSR反应体系
用Arra Yugander[12]设计的100个ISSR引物中筛选到的18条应用于水稻纹枯病菌的引物作为初筛引物,从中筛选出3条重复性好、特异性高的引物进行ISSR-PCR反应,引物序列由Invitrogen公司合成。
将提取好的DNA用上述引物进行PCR扩增,PCR反应程序为94℃预变性4min;94℃变性1min;50℃退火1min;72℃延伸1.5min;35个循环;72℃延伸5min,PCR结束后取5微升PCR产物在含有溴化乙锭(EB)的2.0%琼脂糖凝胶下电泳,使用分子量标记物Takara DL2000 DNA Marker作为标准,将同一条引物扩增且电泳迁移率一致的条带视为同源性条带,即同一位点产物。不同菌株中PCR扩增产生多态性条带,按照有条带赋值1、无条带赋值为0进行统计。应用Popgene软件获得遗传多样性相关指数,生成地理种群间遗传关系,将生成的遗传距离数导入NTsys软件,采用欧式距离加权配对算数平均法建立遗传关系聚类图。
1.6 致病性测定
采用改进后的水稻离体叶片接种方法[13],于水稻6叶期选取完全展开的倒1叶或倒2叶叶片中间8cm部分,接种菌丝块于离体叶片底部,每皿平行等距贴5片叶,重复3个组。另接种无菌菌丝块作为对照。置于28℃,95%RH下培养。48h后测量病斑高度,从而比较致病性差异。
2 结果与分析
2.1 菌株的菌丝融合群
结果(图1)表明94个菌株均属于AG1-IA融合亚群,安徽省水稻纹枯病菌整体构成单一。

图1 水稻纹枯病菌株特异性引物鉴别PCR检测电泳图
2.2 引物筛选及菌株ISSR扩增结果
从18个ISSR引物中选出重复性较好、特异性强的3个引物对94个菌株进行ISSR-PCR分析,扩增位点分别为16、14和21个,扩增的DNA片段大小在100bp到2000bp之间。引物的多态性位点比率均在90%以上,说明供试菌株个体间存在较大的遗传变异。
15个地理种群多态性位点比率在19.61%~84.31%之间,平均为49.67%,种群的多态性位点比率从大到小依次为:AHXN>AHNG>AHWJ>AHCZ>AHCH>AHLJ>AHYS>AHWW>AHJX>AHHX>AHGD>AHFT>AHFY>AHLX>AHBB。表明安徽省纹枯种群之间遗传多样性较为丰富。
2.3 水稻纹枯病菌聚类分析
用NTsys软件对扩增出的多态性条带构建个体间遗传关系UPGMA聚类树状图,结果表明,在相似系数0.65阈值下,可以分成4个遗传聚类群,水稻纹枯病菌间遗传相似系数在0.57~0.96之间,类群1中,望江的全部13株聚类在一起;类群2中,绩溪、和县、颍上、凤台、广德、郎溪和巢湖全部菌株聚类在一起,宁国的9个菌株中8株也聚类在类群2;类群3中,无为和蚌埠全部菌株聚类在一起;类群4较为特殊,只有休宁的一株菌株。这表明来源于同一地区的菌株基本归类在一个类群,表明其与其地理来源存在明显相关性。
2.4 水稻纹枯病菌遗传多样性和遗传分化
水稻纹枯病菌群体的遗传多样性分析结果如表3所示,Nei’s指数H在0.0812~0.2980之间,均值为0.1817;有效等位基因数Ne为1.1386~1.5244,平均有效基因数1.3138,表明该种群内存在明显的遗传分化;Shannon信息指数I为0.1186~0.4425,均值为0.2699,说明具有较高的遗传多样性;以上3个指数的变化规律基本与多态性位点比率变化一致。
总种群基因多样度Ht为0.3048,各个种群内基因多样度Hs平均值为0.1327,种群基因分化系数(Gst)平均值为0.4040(Gst>0.5,种群间的遗传分化明显),说明安徽省不同县市的水稻纹枯病菌种群间存在较为明显的遗传分化,AMOVA分析结果表明,种群间的遗传变异占总变异的27%(P<0.001),种群内遗传变异占总变异的73%,说明安徽省的水稻纹枯病菌种群间变异较为明显,且其遗传变异主要来自种群内部;基因流Nm为0.7378,说明种群间存在一定的基因交流。
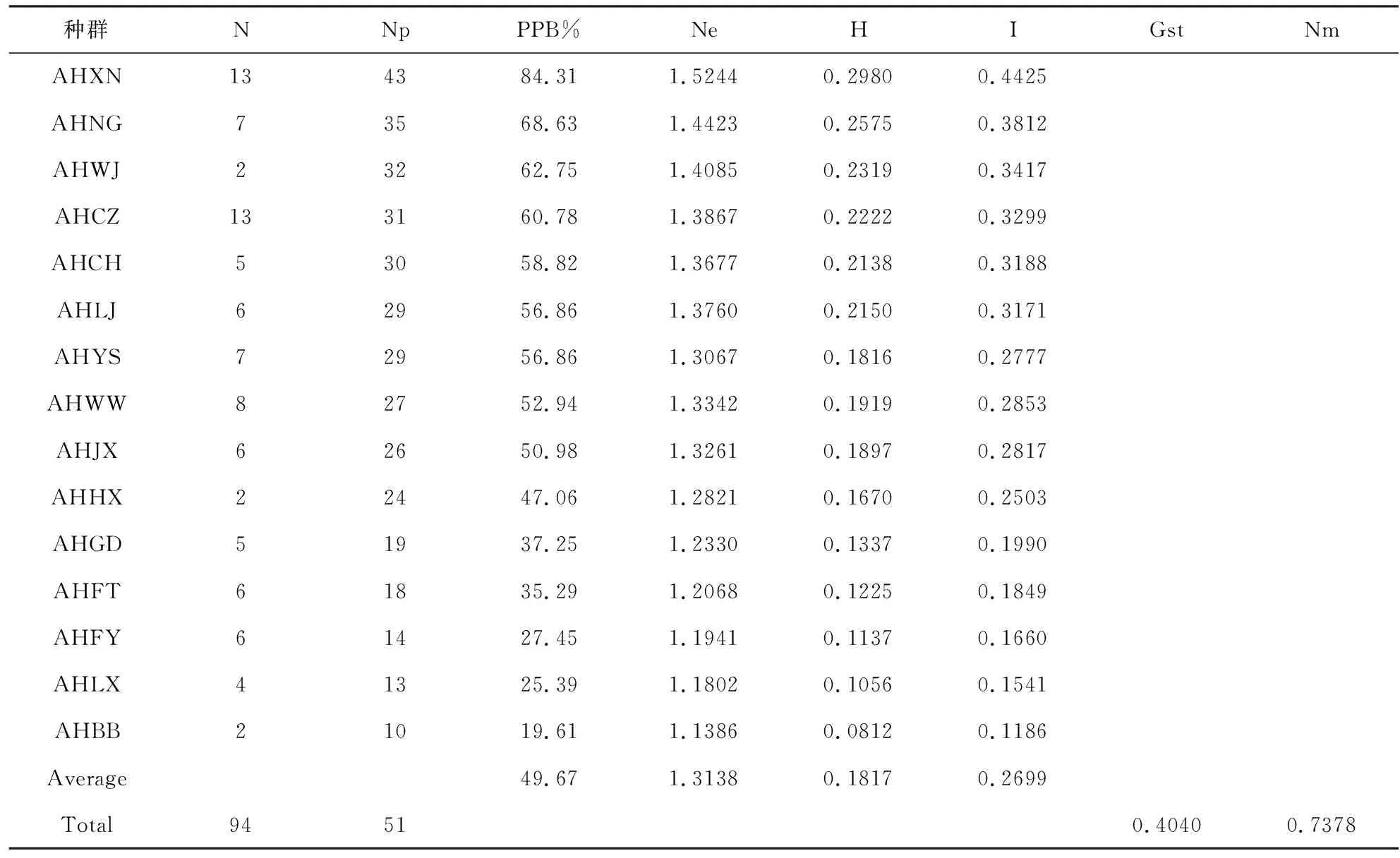
表3 15个不同地理位置水稻纹枯病菌群体的遗传多样性
2.5 病原菌的致病性
水稻纹枯病菌菌株致病性研究结果如表4,可以看出大多数菌株致病性都是中等或较弱;强致病菌株有2株,分别来自和县和无为。各地菌株致病性分布并不均匀,部分地方致病性差异跨度较大。采用Duncan氏新复极差法进行差异性分析,结果表明,各菌株间的致病性存在极显著差异(P<0.01)。
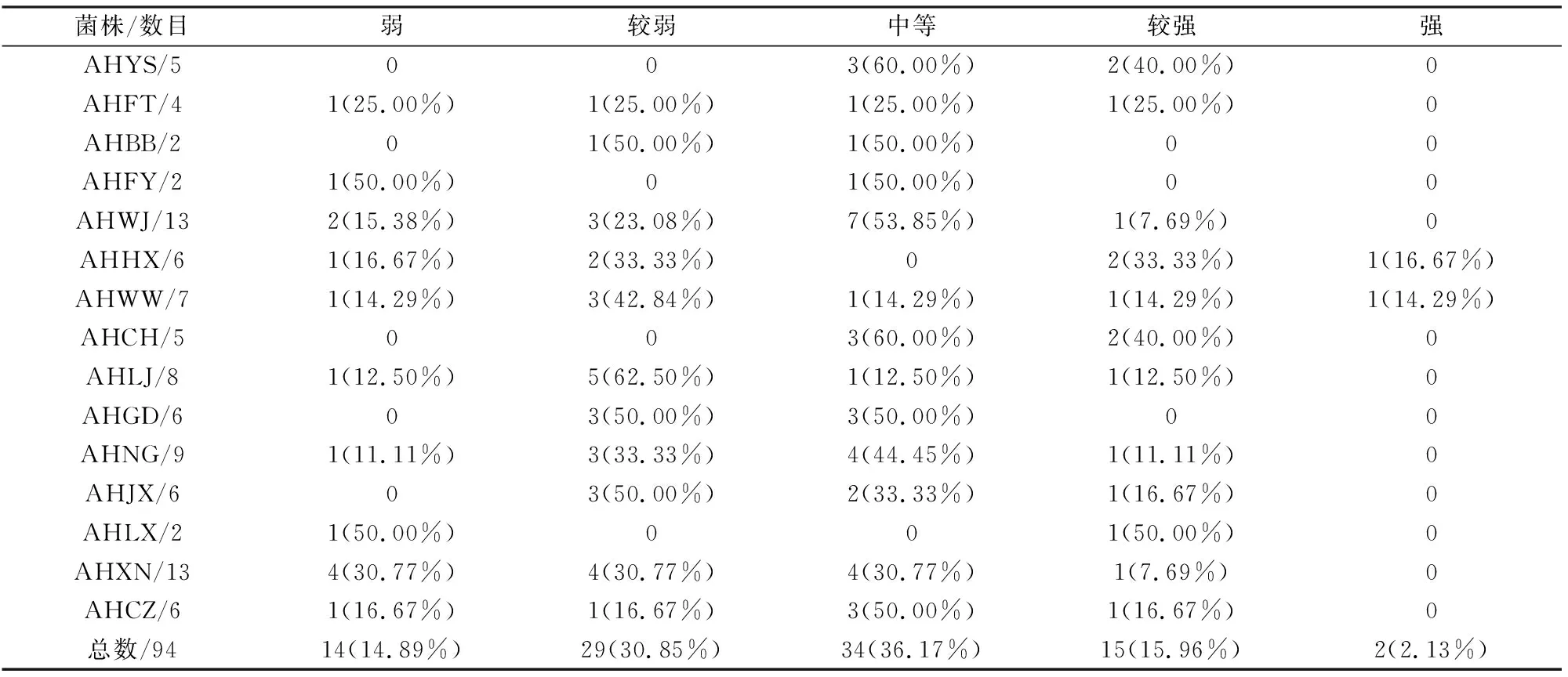
表4 各地水稻纹枯病菌菌株不同致病性k-均值分类结果
3 讨论
3.1 安徽省水稻纹枯病病原菌种类分布
研究所用的94个水稻纹枯病菌菌株来源于安徽省15个代表性县市,相较于黄雯雯等[8]在2010年前采自安徽7个县市的32个菌株而言更加丰富,能够比较全面地反映出安徽省水稻种植区的水稻纹枯病菌的实际情况。特异性引物鉴定技术表明安徽省菌株都隶属AG1-ⅠA融合亚群,与中国东北地区水稻纹枯病原菌融合群不同[14-15]。因此,可以推断安徽省水稻纹枯病菌遗传背景较为单一。
3.2 安徽省水稻纹枯病菌的遗传分布及结合致病性提出防治策略
采用ISSR分子标记技术,本实验反应体系共扩增出51条条带,多次重复试验均得到较好的多态性图谱,结果表明水稻纹枯病病菌具有丰富的遗传多样性。通过NTsys对94个菌株聚类,11个地点的菌株都被聚在同一类中,说明菌株亲缘关系与其地理来源具有明显的相关性,菌株的亲缘关系与地理位置是否相关,其研究结果不尽相同[16-19]。水稻纹枯病菌的致病性在同一融合群内也产生了显著差异,水稻纹枯病菌致病性主要为中等致病性及以上[20],过去大多研究都表明致病性的差异与遗传划分并无显著关联,与本次研究结果相同。研究还发现不同地理位置群体存在一定的遗传差异,尽管种群内部变异占主导地位,但种间的基因交流水平相较前人研究有所增加。发现除仅含休宁1株菌株的类群4以外,其余3个类群中均包含了皖南、皖北及皖中地区的市县,说明安徽省各地水稻纹枯病病菌间基因存在较为频繁的交流。安徽近30年气候研究表明,整体气候逐渐变暖[21],致使水稻种植界限北移,有效积温的增加使得安徽北部也具备水稻种植条件,水稻可种植面积进一步加大。寄主植物的增多,加强了水稻纹枯病在不同地区间的交流。此外,有限的农用机械在被调用的过程中,将不同地区的带有菌核的泥土或附着菌丝的水稻、病草残株带入其他地区,从而使得距离较远的地区菌株间产生交流,也导致了距离较远地区的水稻纹枯病菌种群分子水平上表现出亲缘关系较高的现象。
