煤分子结构模型构建及优化研究
2021-04-17张志军
张志军,王 露,李 浩
(中国矿业大学(北京) 化学与环境工程学院,北京 100083)
0 引 言
在我国能源体系中,煤炭是储量最大的不可再生能源,同时也是重要的基础能源和工业原料[1-2]。正确认识煤的分子结构特征对实现煤炭合理利用及高效转化具有重大意义。 许多现代分析测试技术可用于研究煤的结构特征,如傅里叶红外光谱(FTIR)、X 射线衍射(XRD)、X 射线光电子能谱(XPS)、拉曼光谱、核磁共振碳谱(13C-NMR)和高分辨率透射镜(HRTEM)等。 近半个世纪以来,国内外煤炭研究学者们致力研究煤的结构与性质,发现煤中有大量的亲水基团和缩合芳环,且随着变质程度的升高,煤中芳香环数增加而脂肪结构不断减少[3-5]。 同时,学者们利用化学试验研究和现代测试技术对煤的分子结构进行研究,提出了130 多种煤的大分子结构模型[6],例如,著名的Wiser 模型[7]、Given 模型[8]、Shinn 模型[9]等。 这些结构反映出不同变质阶段的煤分子结构,在一定程度上解释了煤的浮选、溶胀裂解以及气体吸附等行为[10-13]。 随着近几年计算机技术的快速发展,在研究复杂的分子结构及体系时,计算化学对试验的辅助及应用越来越成熟[14-15]。 构建煤的分子模型是对其进行分子模拟的基础,通过分子模拟计算获得煤的合理分子结构可为进一步选择浮选药剂提供理论基础。
以淮北矿业集团青东煤矿煤为研究对象,通过工业分析、元素分析、13C-NMR、XPS 等测试对其分子结构进行分析,建立青东煤的单个大分子结构模型及聚集态结构模型。 为寻找单个煤分子最低能量构型,利用分子模拟软件Materials studio(MS)7.0 对单个分子模型结构进行几何优化及退火动力学模拟研究,并构建出煤的聚集态结构模型,从分子尺度为研究青东煤的浮选药剂吸附机理提供了模型基础。
1 样品及试验方法
1.1 样品采集和制备
煤样采自于淮北矿业集团青东煤矿,采样方法遵循GB/T 482—2008《煤层煤样采取方式》。 在实验室内,按GB/T 478—2008《煤炭浮沉试验方法》对青东煤进行煤岩分离,选取出密度级为-1.30 g/cm3的煤样在70 ℃的条件下干燥至恒重。
1.2 工业分析和元素分析
青东煤样的工业分析和元素分析见表1。 采用德国Vario EL cube 型元素分析仪测定样品的C、H、S、N 元素含量,采用差减法计算O 元素含量。 并根据工业分析和元素分析结果计算青东煤样的氢碳原子比H/C、氧碳原子比O/C、氮碳原子比N/C 和硫碳原子比S/C(表1)。

表1 青东煤样的工业分析和元素分析Table1 Proximate andelemental analysis of Qingdong coal sample
1.3 核磁共振碳谱测试
采用Bruker AVANCE III600 型核磁共振仪对青东煤样进行13C-NMR 分析。 测试参数如下:外径为4 mm 的ZrO2探头,核磁共振频率75.47 MHz,转速7.0 kHz,脉宽4.2×10-6s,采样时间0.05 s,碳氢交叉极化接触时间3 ms,谱宽3 000 Hz,数据采集累加次数6 000 次。
1.4 X 射线光电子能谱测试
采用美国热电Thermo Escalab 250XI 型光电子能谱仪对青东煤样进行XPS 测试。 测试参数如下:单色Al Ka (hv=1 486.6 eV),功率150 W,X 射线束斑500 μm,采用污染碳(284.8 eV)对电荷进行校正,通能窄扫20 eV,宽扫100 eV,真空度为10-8mPa。
2 讨论与结果
2.1 核磁共振碳谱分析
煤的13C-NMR 谱图可划分为3 个区域[16-17]:①化学位移在0 ~80 的脂碳区;②化学位移在80 ~170 的芳碳区;③化学位移在200 左右的羰基碳区。由于煤中官能团种类丰富且结构复杂,13C-NMR 谱图上的峰可由多个子峰叠加形成,因此需要对谱图进行分峰处理,以确定各个官能团的峰位及相对含量[18-20]。 利用Origin8.0 软件对青东煤样的13CNMR 谱图进行分峰拟合,结果如图1 所示。
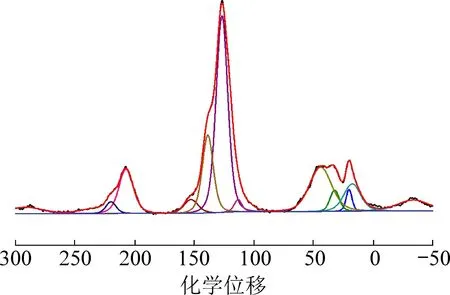
图1 青东煤样的13C-NMR 分峰拟合Fig.1 13C-NMR spectra of Qingdong coal
根据青东煤中各官能团的峰位及其相对百分含量,计算出煤样的12 个重要结构参数。 由于受核磁测试试验边带效应的影响,青东煤样的羰基碳区整体移至215 左右,作用强度比实际偏大[21]。 研究发现,煤化程度越高,羰基峰强度越偏离实际强度,将对煤炭芳香率与脂碳率的计算产生不可忽视的误差。 为降低该种误差,对青东煤样的芳香率f′αl与脂碳率fαl进行修正[22-23],结果见表2。
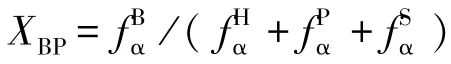

表2 青东煤样的结构参数Table 2 Structure parameters of Qingdong coal
注:fα为sp2 杂化碳;fαl为sp3 杂化碳;fCα为与羰基相连的芳香碳;f′α为芳香碳;fHα为质子化芳碳;fNα为非质子化碳;fPα为与氧原子相连的芳碳;fSα为被烷基取代的芳碳;fBα为桥接芳碳;f*αl为脂肪烃中的甲基;fHαl为亚甲基、季碳或次亚甲基;fOαl为与氧原子相连的脂肪碳。
2.2 X 射线光电子能谱分析
煤样中碳、氧、氮、硫以多种官能团形式存在,其XPS 谱图经多个子峰叠加形成。 采用Case XPS 软件对4 个XPS 谱图进行分峰拟合,依据各结合能的归属,确定它们在样品中的存在形式[26-29]。 青东煤的碳原子在结构中以4 种形态(图2 和表3)存在:①284.63 eV 峰归属于芳香石墨化碳(C ═C);②285.07 eV峰归属于脂肪族碳(C—H);③285.46 eV 峰归属于羟基结构(C—O)和羰基结构(C ═O)。 计 算 子 峰 的 相 对 面 积, C ═C 结 构 碳 占65.15%,即青东煤样中的碳主要以芳香结构碳为主,与核磁共振碳谱图结果一致。 氧原子在青东煤表面结构中以2 种形态(图3 和表4)存在:①532.08 eV 峰为羟基氧(C—O)或醚氧(—O—),子峰相对面积45.39%;②533.20 eV 和533.94 eV 峰均为羰基氧(C ═O),子峰相对面积54.61%。 由此可知,青东煤样中的氧原子以羰基和酚羟基2 种形式存在。 氮原子在青东煤中的存在形式为吡啶和吡咯(图4 和表5):①398.64 eV 峰为吡啶型氮(N—6),相对面积为59.48%;②400.49 eV 峰为吡咯型氮(N—5),相对面积40.52%。 硫原子在煤样中以亚砜型硫化物和噻吩型硫化物形态存在。 165.28 eV 峰为亚砜型硫化物,164.16 eV 峰为噻吩型硫化物(图5 和表6)。

图2 青东煤样的XPS C(1s)谱图Fig.2 XPS C(1s) spectra of Qingdong coal

表3 青东煤样的XPS C(1s)数据Table 3 XPS C(1s) data of Qingdong coal
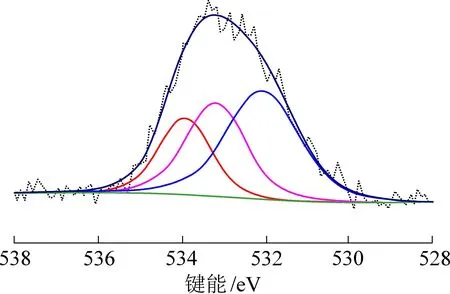
图3 青东煤样的XPS O(1s)谱图Fig.3 XPS O(1s) spectra of Qingdong coal

表4 青东煤样的XPS O(1s)数据Table 4 XPS O(1s) data of Qingdong coal

图4 青东煤样的XPS N(1s)谱Fig.4 XPS N(1s) spectra of Qingdong coal

图5 青东煤样的XPS S(2p)谱Fig.5 XPS S(2p) spectra of Qingdong coal

表5 青东煤样的XPS N(1s)数据Table 5 XPS N(1s) data of Qingdong coal

表6 青东煤样的XPS S(2p)数据Table 6 XPS S(2p) data of Qingdong coal
3 青东煤分子结构及模型构建
3.1 芳香结构
文献[30-31]研究表明,当煤中碳质量分数为81%~91%时,芳香环平均环数为2 ~5 个。 青东煤样的碳质量分数为90.44%,XBP为0.35,2 环芳香化合物萘的XBP为0.25,3 环的芳香化合物蒽的XBP为0.4[32],通过调整芳香环的类型和数量,使结构模型中的XBP接近0.35。 当青东煤结构中萘的数量为2个,蒽为4 个时,结构中的XBP=0.357。 基于ACD/CNMR Predictor 软件只能用于不超过255 个原子(不包括H 原子数目)的分子,因此模型的芳香结构类型和数量见表7, 其中芳香碳原子数量为97 个[33]。

表7 青东煤样化学结构模型中芳香结构单元的类型Table 7 Types of aromatic structure unit in chemical structural model of Qingdong coal
3.2 脂肪结构

3.3 杂原子结构
由上可初步确定青东煤样结构的碳原子总数为150,结合元素分析中各元素与碳元素原子比可推算出煤样模型中氧、氮、硫原子数量分别为3、2、0。 研究表明,煤中含氧官能团包括甲氧基、羧基、羟基和羰基等[37]。 随着煤化程度的增高,甲氧基含量迅速减少,在硬褐煤中基本消失;羧基主要存在于褐煤中,而羟基和羰基存在于整个烟煤阶段[38]。 结合13C-NMR 及XPS 分析结果可确定青东煤结构模型中3 个氧原子的存在形式为2 个羰基和1 个酚羟基。
吡啶型氮和吡咯型氮是煤结构中氮的主要存在形式[39]。 结合XPS 结果可知,青东模型中的2 个氮原子分别以吡啶型氮和吡咯型氮的形式存在。 XPS结果还表明,煤样中的有机硫包括亚砜型和噻吩型硫化物。 由于该煤样中硫质量分数仅为0.26%,计算得出的硫原子个数不足1 个,且模型中原子数量有限,因此构建模型时不再考虑硫原子。
3.4 青东煤分子结构模型
采用分子结构绘图软件Chem draw/Chem3D 构建青东煤平面结构模型。 应用ACD/CNMR Predictor软件计算出模型结构的化学位移,将数据导入gNMR 软件获得模型的计算13C-NMR 谱图[40]。 利用Origin8.0 软件将模型的计算13C-NMR 谱图与试验测试谱图数据进行匹配,通过调整模型中的各个结构单元的连接方式,对结构模型进行修正[41]。 青东煤结构模型计算13C-NMR 谱图和试验13C-NMR谱图对比效果如图6 所示。
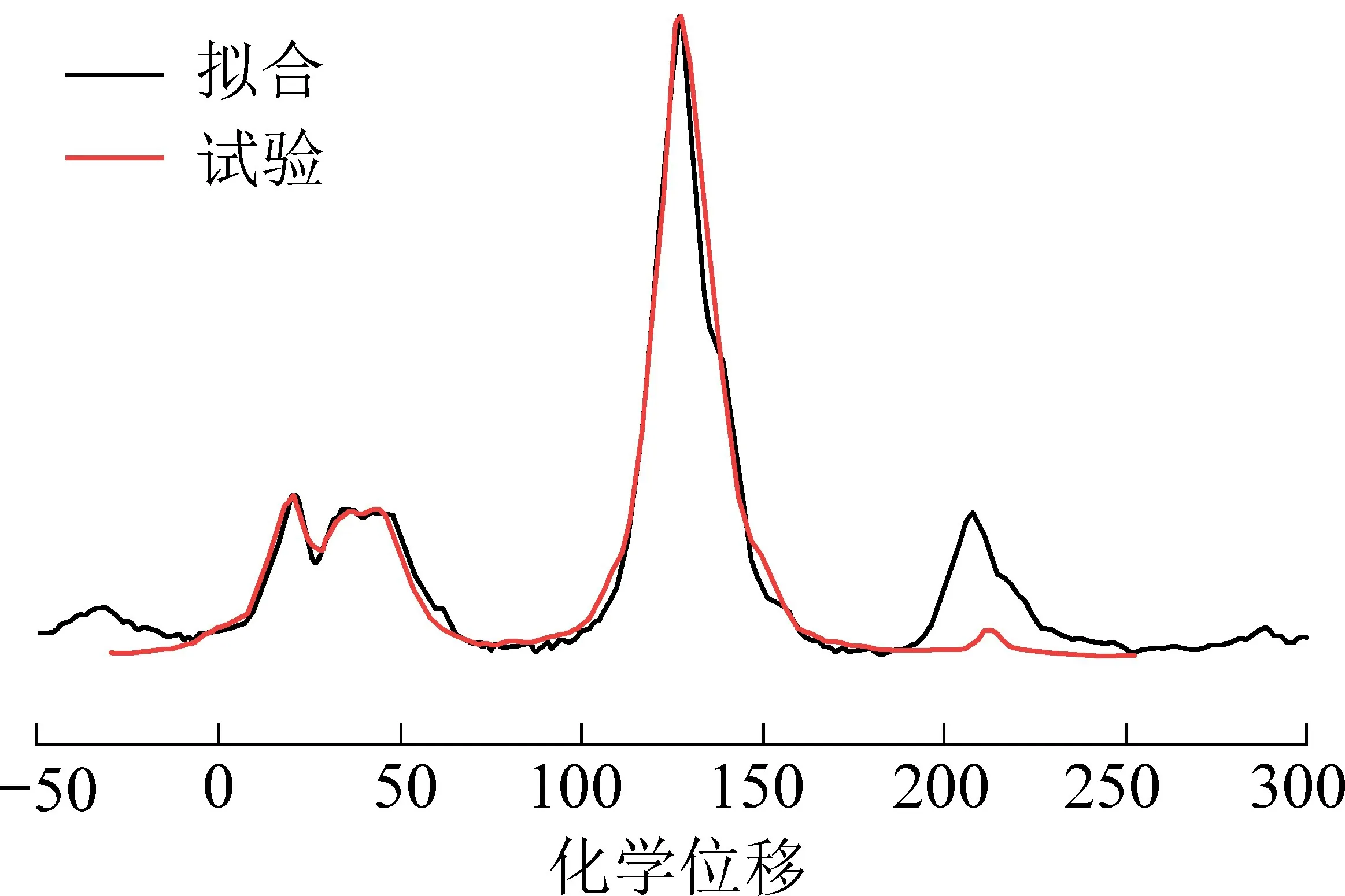
图6 青东煤的结构模型计算13C-NMR和试验13C-NMR 谱Fig.6 Experimental and calculated13C-NMR spectrum of Qingdong coal
由图6 可知,模型计算谱图和试验谱图整体吻合较好,但由于在核磁共振碳谱测试试验过程中存在边带效应,使实验谱图中的碳氧区吸收峰强度偏大,因此在化学位移200~230 区域计算谱图吸收峰强度明显低于试验谱图。 青东煤分子结构的分子式为C142H128N2O3,其分子量为1 910.60。 碳、氮及氧元素质量分数分别为89.27%、1.47%、2.51%,与元素分析中各元素相对百分含量接近;而结构中的氢质量分数为6.75%,与元素分析中的4.96%偏差较大,这是因为在构建煤结构时未考虑硫元素造成的比例偏差。

表8 青东煤样结构模型的结构参数Table 8 Structure parameters of macro-molecular structure model of Qingdong coal
综上所述,构建的青东煤分子平均结构模型结果与元素分析、XPS 分析及13C-NMR 分析结果吻合度较好,可反映出青东煤的平均分子结构特征。
4 单个分子结构模型的分子模拟
4.1 单个分子结构模型
为确定单分子结构模型的最低能量构型,利用MS 软件对青东煤单个分子结构模型进行几何优化及退火动力学模拟。 将青东煤大分子结构(图7)导入MS 软件,使其结构饱和加氢。 几何优化采用Forcite 模块,优化方法为Smart Minimizer 方法。 参数设置如下:基于原子的总能量计算库仑能和范德华能,原子的净电荷采用电荷平衡法获得[42],迭代步数为5 000 步,收敛标准采用Fine。 几何优化后,继续采用Forcite 模块的Anneal 进行高温弛豫。 模拟过程在NVT 系综下进行,选择Nose 控温法;初始温度设为300 K,最高温度设为800 K;升温速率为60 K/次,模拟时间为1 fs;每次循环结束后对输出结果再次进行分子力学优化,其设置参数如前所述。该力场计算定义[43]为
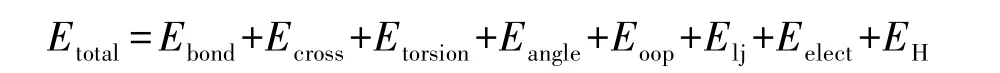
其中:Ebond为键伸缩能;Ecross为交叉项相互作用能;Etorsion为二面角扭转能;Eangle为键角能;Eopp离平面相互作用项;Elj为范德华能;Eelect为静电相互作用;EH为氢键作用能。 其中Ebond、Ecross、Etorsion、Eangle、Eopp为价电子能;Elj、Eelect、EH为非成键能。
选取模拟结果中能量最低的结构作为青东煤的最优几何构型,如图8 所示。 可以看出,经过分子力学和分子动力学优化后,单个分子结构为达到空间结构上官能团之间的最小斥力,部分桥键、脂肪键等化学键发生了明显的扭转[44]。 由于芳香环之间π-π相互作用,相邻芳香片层之间趋于平行排列,层间距增大,呈现出明显的立体感[45]。
表9 是青东煤结构模型优化前后的能量组成,优化前总能量为2 121.14 kJ/mol,优化后为1 255.85 kJ/mol,总能量大幅度降低。 优化后的模型中,键伸缩能及范德华能降低,其他项均升高,而总能量明显降低,说明该结构中的键伸缩能及范德华能占主导地位,其中键伸缩能属于价电子能,范德华能属于非成键能。 由于煤是由多个芳环组成的大分子结构,在模拟的过程中,青东煤分子模型由二维的平面结构转化为三维的立体结构,分子结构中部分化学键扭转,键角变化,造成键角能及扭转能增大,同时引起键伸缩势能的降低[46]。 优化后的立体结构中,芳香片层之间的平行排列产生较大的色散力,是范德华能下降的主要原因[47]。
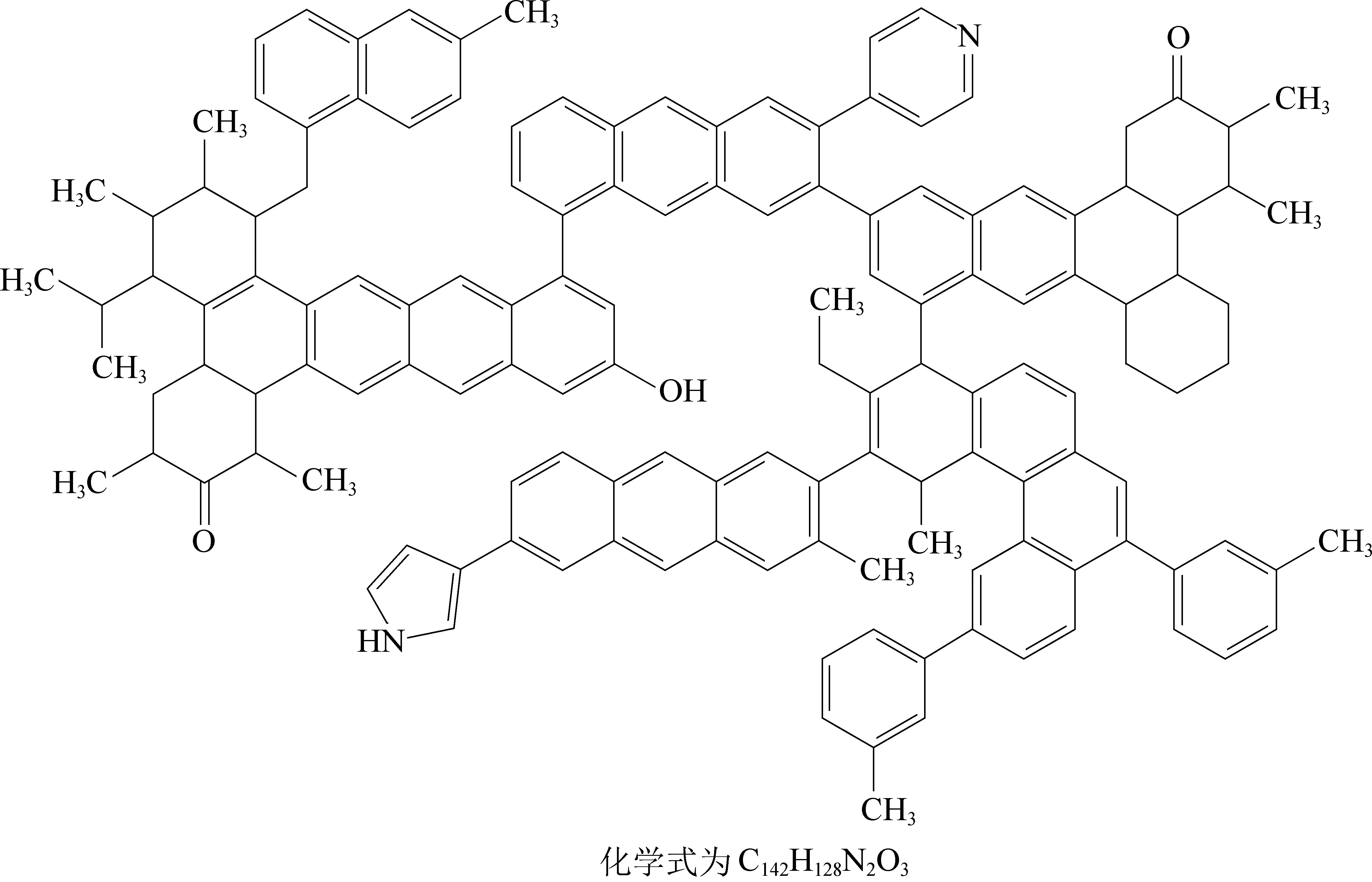
图7 青东煤平面分子结构模型Fig.7 Plane model molecular structure in Qingdong coal

图8 单个煤分子的最低能量结构模型Fig.8 Energy-minimum conformation of a single coal model
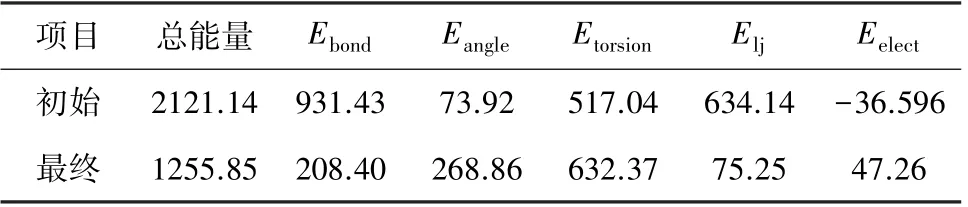
表9 青东煤结构模型的能量组成 kJ/molTable 9 Potential energy for conformation of Qingdong coal
4.2 聚集态结构模型
利用MS 软件中的Amorphous Cell(AC)模块将18 个优化后的青东煤大分子随机添加到尺寸为40Å×40Å×40Å(1Å =0.1 m)的晶胞中,并添加三维周期性边界条件[48]。 设定结构模型的密度为1.25 g/cm3。 首先利用分子力学进行几何优化,然后进行退火动力学模拟对该体系进行弛豫,温度设置为298~1 098 K,并使最终温度保持在298 K[49-50]。 选定能量最低的构型体系作为周期性边界条件下青东煤分子聚集态结构模型,如图9 所示。 从聚集态结构模型中可以看出,经过分子力学和分子动力学模拟优化后,分子结构发生明显弯曲、扭转。 由于煤是非晶体物质,无长程周期性,受周围分子的制约,原本近似平香碳结构发生扭曲形变,片层结构行排列的片状芳杂乱,使整个凝聚态结构模型更加紧凑[51]。 从图9 知青东煤大分子分布较为均匀,表面含氧官能团分布均匀,可反映出青东煤样表面的结构特征。

图9 青东煤聚集态结构模型Fig.9 Aggregation-state for Qingdong coal molecules
5 结 论
1)根据青东煤的核磁共振碳谱得出表征煤结构的12 个结构参数,并进行修正;根据参数计算出煤分子结构中芳香桥碳与周碳比为0.35。13C-NMR及XPS 结果表明,青东煤的碳原子在结构中主要以芳香石墨化碳结构为主,其XPS 谱图子峰相对面积占65.15%;煤样中芳香结构单元包括2 个苯环,2个萘,4 个蒽;煤分子结构中的杂原子别以2 个羰基和1 个酚羟基、1 个吡啶和1 个吡咯存在。
2) 利 用Chem draw/Chem3D 及ACD/CNMR Predictor 构建了与青东煤的13C-NMR 试验谱图拟合较好的结构模型;最终构建的青东煤单分子结构分子式为C142H128N2O3,分子量为1 910.60。
3)利用MS 软件对单个分子结构模型进行几何优化和分子动力学模拟后,桥键、脂肪键等化学键发生了扭转,分子内芳香片层之间的π-π 相互作用使相邻芳香片层之间趋于近似平行排列;优化前后分子总势能由2 121.14 kJ/mol 下降到1 255.85 kJ/mol,键伸缩能及范德华能占主导地位。
4)构建出聚集态结构模型,经过分子力学和分子动力学模拟优化后,聚集态结构模型中的大分子受周围分子的制约,原本近似平行排列的片状芳香碳结构发生扭曲形变,片层结构杂乱,整个凝聚态结构模型紧凑;构建的聚集态结构模型可为后续探索浮选药剂吸附模拟提供模型基础。
