PI3Kδ过度活化综合征诊疗进展*
2021-04-14杨夏影马银娟潘耀柱
杨夏影,马银娟,潘耀柱
(中国人民解放军联勤保障部队第九四〇医院 血液科,甘肃 兰州730050)
PI3Kδ 过度活化综合征(activated PI3 kinase delta syndrome, APDS)是由基因突变导致的p110δ 过度活化,从而导致T 细胞和B 细胞发育和功能改变为特征的联合免疫缺陷综合征。该病于2013年由ANGULO等[1]首次报道。现已发现PIK3CD(编码PI3K 催化亚基P110δ)及PIK3R1基因(编码PI3K1A 类调节亚基P85α)功能获得性突变可致APDS,分别称为APDS1和APDS2[2-3]。该病临床表现多样,常见有反复呼吸道感染、支气管扩张、自身免疫性血小板减少、非肿瘤性淋巴组织增生、淋巴瘤、巨细胞病毒(CMV)和/或EB 病毒血症等,好发于婴幼儿(0.58~3.00 岁),男性多于女性(男/女≈1.4),病死率约为12.1%[4]。
1 APDS的流行病学特征
JAMEE 等[4]将2013~2018年发表的243 例APDS患者(男性123 例,女性87 例,性别未知33 例)进行系统综述,中位确诊年龄为12.0(6.5~21.5)岁,中位确诊延迟为7.0(3.4~14.0)年;88 例(38.6%)患者有免疫缺陷家族史。在可追踪到的患者中,大部分尚存活,少数已病故,恶性肿瘤(如淋巴瘤和急性髓系白血病)、心脏骤停、肠穿孔引起的严重感染性休克及多器官功能衰竭为病死的主要原因。
2 APDS主要分子发病机制
2.1 PI3K信号通路与APDS
磷脂酰肌醇3-激酶(PI3Kδ)蛋白家族参与细胞存活、生长、代谢和能量稳态等多种细胞功能的调控。IA 类PI3K 分子是由p110 催化亚基(p110α、p110β 或p110δ)和调节亚基(p85α、p55α、p50α、p85β 或p55γ)组成,后者影响催化亚基的定位和激活。在哺乳动物中,p110α 和p110β 普遍表达,而p110δ 是免疫细胞中最主要和最丰富的亚型,其活化和功能缺失将对免疫系统发挥重要作用[5]。
PI3Kδ参与PI3K-AKT-mTOR信号通路(见图1)[6],PI3Kδ 促使磷脂酰肌醇-3,4-二磷酸(PIP2)转化成磷脂酰肌醇-3,4,5-三磷酸(PIP3),随之使Akt 磷酸化。在淋巴细胞中,活化的Akt 使众多底物磷酸化:①FOXO(叉头盒)转录因子如FOXO1,对淋巴细胞发育、活化和迁移的重要基因表达具有调控作用。②mTOR 是一种保守的丝氨酸/苏氨酸激酶,包含两种复合体:mTORC1 和mTORC2。mTORC1 通过影响下游分子磷酸化,如翻译延伸因子4E-BP 和核糖体S6 激酶,调节淋巴细胞生长、代谢、遗传、增殖、存活、蛋白质翻译和转录[7];mTORC2 则促进淋巴细胞中Akt 的完全磷酸化和激活[8]。③糖原合成酶激酶3β(GSK3β)[8]、转录因子BACH2[9]做为重要的Akt 下游分子在淋巴细胞发育中也发挥重要作用。此外,PTEN 和SHIP-1/2 是PI3K 信号通路中关键的PIP3 磷酸酶,与PI3K 的功能恰恰相反,其可通过去磷酸化将PIP3 转变为PIP2,进而减少Akt 的活化,阻止由Akt 调控的下游信号传导,对维持PI3K 信号平衡具有重要作用。PIK3CD和PIK3R1基因突变导致PI3K 信号通路过度活化,均可促使p110δ 对质膜的亲和力增加,从而导致PIP2 转化成PIP3 增多,导致下游通路Akt 和mTOR 过度活化,过度活化的AKT 将抑制下游众多信号分子的正常表达。在长期高活性PI3Kδ 下,导致PI3K 信号传导与细胞功能平衡紊乱,T 细胞过度活化、耗竭、衰老及B 细胞发育有缺陷、抗体产生减少等,表现为以感染、B 细胞淋巴瘤等易感性增加为特点的细胞免疫、体液免疫功能缺陷。总之,PI3K 信号通路对调节淋巴细胞发育和免疫反应有广泛的多效性影响。
2.2 APDS中B细胞改变
在B 细胞中有多种受体可招募PI3K,如共同受体CD19 的B 细胞抗原受体和磷脂酰肌醇3-激酶的B 细胞适配器。此外,PI3K 也可被Toll 样受体、趋化因子CXCR5 和细胞因子(IL-4、IL-21、B 细胞激活因子)等激活[10]。PI3K 在B 细胞信号传导过程中的重要性不言而喻。当PI3Kδ 功能获得性突变时可导致在BM 中B 细胞发育异常,未成熟B 细胞比例增加,成熟循环B 细胞减少。虽然APDS 患者的血液循环B 细胞比例正常,但过渡期B 细胞百分比增加,幼稚和记忆B 细胞减少[1]。此外,观察到非类别转换(IgD+) 和类别转换记忆B 细胞(IgG+和IgA+)群体减少[11],表明记忆形成和同型转换都存在缺陷。长期抗体反应的产生在很大程度上是通过生发中心(GC)的发育介导的,GC 是B 细胞对来自T 细胞和抗原的信号作出反应而发生体细胞超突变并产生高亲和力抗体、记忆B 细胞和长寿浆细胞的部位[12]。有趣的是,在与人类数据相一致的Pik3cdE1020K/+小鼠中[13],有学者观察到尽管分离的淋巴组织中的GC B 细胞增多,而血液中滤泡辅助性T 细胞(Tfh)的比例更高,但其对肺炎球菌多糖疫苗和接种B 型流感嗜血杆菌疫苗的反应却较差。此外,有文献报道称,FOXO1 对调节BM 和外周B 细胞分化多个步骤的基因表达至关重要,例如IL-7 受体、重组激活基因(RAG1、RAG2)、IKAROS、活化诱导的胞苷脱氨酶、CD62L 和BLNK,可能有助于改变B 细胞的发育[14]。在APDS 患者中易观察到非肿瘤性淋巴组织增生,如淋巴结病、脾肿大、肝肿大以及EB 病毒相关性淋巴瘤的发生。在10~14 个月的Pik3cdE1020K/+小鼠中观察到约20%小鼠出现自发的癌前病变和/或肿瘤。APDS 患者肿瘤的增加可能由于淋巴细胞的增殖能力增强、代谢改变和/或细胞死亡减少,导致癌基因和/或肿瘤抑癌基因突变的积累。此外,T 细胞对肿瘤的无效监测也有助于癌细胞的生长[6]。
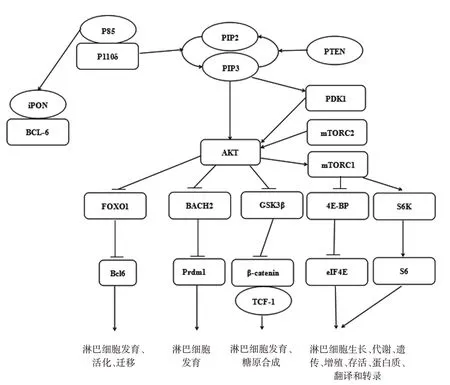
图1 PI3K信号通路与淋巴细胞
2.3 APDS中T细胞改变
活化的PI3Kδ 如何促进效应细胞分化?首先,几个由PI3Kδ 活性控制的转录调节因子对调控效应T 细胞与记忆T 细胞之间的平衡至关重要。失活的FOXO1 抑制记忆细胞并促进效应CD8+T 细胞形成;BACH2 抑制Prdm1 和Bcl-6,促进多种效应细胞谱系的产生并减少调节性T 细胞的形成[15];其次,活化的PI3Kδ 可能影响调节途径T 细胞代谢。激活的T 细胞转换为具有特定特征的有氧糖酵解,增加葡萄糖摄取和糖酵解酶的表达。正常情况下,活化的CD4+T 细胞在离开次级淋巴组织后,进入到外周血中,从而形成记忆T 细胞。此时在部分T 细胞上仍有CXCR5 的表达,这种经过抗原刺激后发育的T细胞称为效应T 细胞,该细胞归巢至B 细胞滤泡,在IL-21、诱导T 细胞共刺激分子及CD40L 等相关的细胞分子参与下,完成其辅助B 细胞的使命。如上文所述,长期抗体的产生在很大程度上依赖于GC的发育,而Tfh 是近年来发现的CD4+T 细胞新亚群,其关键驱动并维持GC 的形成[16]。在Pik3cdE1020K/+小鼠中,尽管Tfh 细胞和GC 有所增加,但研究发现循环滤泡辅助性T 细胞细胞的主要成分是CXCR3+,这一亚群的Tfh 细胞提供幼稚B 细胞的帮助不足[17]。PI3K/Akt 途径驱动多种趋化因子和归巢受体的表达[18],在APDS 患者中,Tfh 细胞可能无法归位或无法正常运行,这可能会导致高度混乱的GC,尤其是在慢性刺激的情况下。而且GC 的结构杂乱无章,亮区(LZ)与暗区的比率失衡。此外,Tfh 细胞不再局限于LZ,而分布于整个GC[13]。Tfh 细胞数量的增加,本身也损害GC B 细胞选择和亲和力成熟[19]。除此之外,目前已经证实在记忆性B 细胞与浆细胞的形成过程中,Tfh 起到非常关键的作用,但具体作用机制尚不清楚。另外,从免疫表型上讲,APDS 患者中虽然CD8+效应T 细胞增加,但其效应功能是有缺陷的,导致无法控制慢性病毒感染,包括CMV 和EBV 感染[2];T 细胞效应功能受损可能是由于T 细胞衰老和T 细胞耗竭,CD8+T 细胞失去再生能力和IL-2 分泌,随后减弱效应细胞因子如肿瘤坏死因子(TNF-α)和干扰素(IFN-γ)的分泌,最终其对凋亡变得敏感,导致这些细胞损失[20]。同时,这些细胞上调了几种抑制性受体的表达,包括程序性死亡受体1(PD-1)、CD160 和CD244[21]。因此,活化的PI3Kδ可引起多种辅助、效应T细胞的功能障碍。
3 APDS的临床表现
APDS 严重、复杂的细胞免疫和体液免疫缺陷,在机制上解释了其多样性与异质性的临床表现,如反复呼吸道感染、支气管扩张、非肿瘤性淋巴组织增生、自身免疫性疾病、淋巴瘤、CMV 和/或EB 病毒血症、神经发育和生长发育延迟等。轻者在成人期尚无相关症状,重者在儿童期可表现出致死性严重的免疫缺陷,且并发恶性肿瘤的风险高[22]。
3.1 感染
在已报道的APDS患者中,普遍有反复呼吸道感染史,其可能为该病的唯一表现,或表现为反复而严重的感染症状。COULTER 等[23]报道了53 例APDS1患者,有51 例(98%)表现为反复呼吸道感染,其中影像学检查确诊的肺炎85%,反复中耳炎49%(造成永久性听力丧失占8%),慢性鼻窦炎(45%),扁桃体炎(28%)。相比之下,在36 例APDS2 患者中,ELKAIM 等[2]描述了上呼吸道感染(既有中耳炎又有鼻窦炎100%),下呼吸道感染(既有支气管炎又有肺炎70%)。由于免疫功能缺陷,APDS 患者易感染各种病原体,病毒感染最为常见,如EB 病毒、CMV、水痘-带状疱疹病毒、人乳头瘤病毒;细菌感染常见于肺炎链球菌、流感嗜血杆菌、金黄色葡萄球菌以及真菌感染(白色念珠菌)等也较多见[4]。
3.2 非肿瘤性淋巴组织增生
JAMEE 等[4]分析文献报道的243 例APDS 患者(见表1),其中APDS1有179例,APDS2有64例。淋巴结肿大是常见的淋巴组织增生性疾病,其次为脾肿大和肝肿大。淋巴结肿大的APDS 患者有149 例,其中APDS1 有100 例(55.9%),APDS2 有49 例(76.6%)。可见淋巴结肿大好发于APDS2。然而,胃肠道和呼吸道的良性淋巴组织增生常见于APDS1,部分APDS2可见扁桃体肿大。

表1 APDS临床表现谱 例
3.3 淋巴瘤
JAMEE等[4]报道的31例(12.8%)发展为恶性肿瘤性疾病,多见于青少年。其中弥漫性大B 细胞淋巴瘤14例,9例为经典型霍奇金淋巴瘤,边缘区B细胞淋巴瘤5 例,多发性淋巴瘤8 例。有慢性病毒感染或既往有病毒感染史与无病毒感染史患者相比,前者恶性肿瘤性疾病的患病率更高。既往有EB 病毒感染史的67例患者,其中12例发展为淋巴瘤。在27例淋巴瘤患者中,12 例有EB 病毒感染史。可见有EB 病毒感染史者其患淋巴瘤的风险显著升高[4]。有文献报道1 例既往有EB 病毒感染史且以霍奇金淋巴瘤(HL)及嗜血细胞性淋巴组织增生综合征(HLH)为临床表现的APDS 患儿,在最初的6年里,由于病情相对无害而未能被确诊。故在诊断HL 和/或HLH 时应高度警惕APDS[24]。
3.4 自身免疫性疾病
大约25%APDS 患者有自身免疫性或炎症性疾病的临床表现。由于多脏器均可受累,故其临床表现多样。如与免疫相关的溶血性贫血、血小板减少性紫癜、甲状腺疾病、类风湿性关节炎、系统性红斑狼疮(SLE)、皮肌炎、炎症性肠病和肾小球肾炎等。其中自身免疫性溶血性贫血、原发免疫性血小板减少症最为常见。部分患者可见炎症性肠病,少数患者有多器官自身免疫性病变以及SLE[4]。
3.5 其他表现
支气管扩张是28%~29%患者最为常见的结构性并发症,其中约半数有反复肺炎病史。尽管肺炎好发于APDS2 患者,但APDS1 患者的支气管扩张更为常见。大约21% 患者有发育不良表现,APDS2 的发病率更为显著,可能与PIK3R1基因杂合突变导致的SHORT 综合征[身材矮小、关节和/或(腹股沟)疝过度松弛、视觉障碍、Rieger 异常和出牙延迟]相关[25]。部分患者有中耳炎和扁桃体炎。精神或学习障碍在APDS 患者中也时有报道(14%),其在APDS2 患者中较为多见[4]。
4 实验室特征及影像学表现
4.1 免疫学特点
APDS 的免疫学特征主要为CD4+T 淋巴细胞减少,效应/效应记忆CD8+T 淋巴细胞增加,过渡B 淋巴细胞增加;而免疫球蛋白水平是多变的,有部分患儿存在IgG 降低,低IgA 和高IgM 也较常见[23]。成熟B 细胞降低、类别转换的记忆B 细胞减少以及衰老T 细胞增加也有报道[3]。此外,有文献报道称过度活化、耗竭和衰老的T 细胞可共存于APDS 患者中,在患者体内T 细胞的活化指标HLA-DR、CD38,T 细胞耗竭或免疫抑制指标PD-1、NKG2D,T 细胞衰老指标CD57 的表达均升高[26]。
4.2 PIK3CD与PIK3R1基因分析
自首次发表以来,在PIK3CD与PIK3R1基因中通过全外显子测序技术已至少发现13 个突变位点(11个激活PIK3CD基因的错义突变,1个剪接位点和1个PIK3R1基因的错义突变)可致APDS(见表2)[4]。数据显示,c.3061 G>A (p. E1021K)位点突变在APDS1 为85%,在APDS2 中,c.1425+1 G>(A,C,T)(p.434-475del)位点占79%,以前者突变最为普遍。在世界各地不同国家、部分民族中均发现了APDS 的散在病例,但现有文献均未提及基因突变与种族背景的相关性,具体情况有待研究。由于APDS 是一种罕见的高外显率单基因疾病,在大量健康受试者中似乎不太可能发现这些突变体。因此,排除在健康人群中检测到的突变形式,如在gnomAD 数据库中报告的信息,将有助于初步筛查潜在的相关突变[27]。在一些家系中发现APDS 的儿童基因突变表现为新发,而在他们的父母中并不存在,并且在E1021K 突变的家庭中进行长期单倍体基因型分析显示没有创始人效应[28]。PI3Kδ 的激活可能在配子形成过程中为细胞提供选择性优势以及是否存在基因开关等假说有待进一步探索。

表2 APDS基因突变谱
目前早期行免疫相关基因筛查,Sanger 验证可缩短确诊时间。有研究报道在部分患儿的PI3K 信号通路中一个抑制分子(PTEN)的失功能突变可导致与APDS 临床、免疫表型类似的免疫缺陷[29]。故在诊断与APDS 相似特征的未确诊患儿,应积极筛查与PI3K 通路过度活化相关的其他致病基因。
5 APDS的诊断
具有反复呼吸道感染、非肿瘤性淋巴组织增生、淋巴瘤、CMV 和/或EB 病毒血症、高IgM 血症等免疫缺陷相关临床表现的初发患者,如婴幼儿、青少年及少数成年患者;病程迁延不愈或无症状病程的青少年、部分成年患者以及有免疫缺陷病家族史者均应考虑本病,同时应早期行基因二代测序技术筛查确诊,并与其它原发性免疫缺陷病,如高IgM 综合征、X 连锁淋巴组织增生症、普通变异型免疫缺陷等[22]相鉴别。
6 APDS的治疗
传统治疗,如预防性抗菌药物、免疫球蛋白替代(IRT),但其疗效欠佳,病死率较高,对传统治疗无效或疗效欠佳且具有严重和复杂临床表现的APDS 患者可考虑行造血干细胞移植(HSCT)。随着对该病的PI3K 信号通路的深入研究,近年来发现了新的靶向药物。目前有2 种药物,一种是抑制PI3K信号通路下游:雷帕霉素;另一种是p110δ 小分子抑制剂:leniolisib(CDZ173)、Nemiralisib(GSK2269557)。目前临床研究发现,阻断PD-1 与PD-L1 的相互作用在治疗反复或持续性病毒感染方面可能是一个新的靶点[21]。
6.1 抗菌药物预防
对APDS 的队列研究进行分析,有一半以上的APDS 患者进行了预防性抗生素应用。抗菌药物的预防性应用不但可满足部分患者需求,与IRT 的联合应用在预防患有支气管扩张症患者的呼吸道感染方面尤为重要。相比之下,预防性抗病毒和抗真菌药物的应用则较为少见。此外,在接种过牛分枝杆菌卡介苗(BCG)疫苗的APDS 患者中,报告持续的卡介苗局部皮肤反应[2,23]。CHIRIACO 等[30]报道,APDS 患者单核细胞来源的巨噬细胞在体外未能消除BCG 感染,这就提出APDS 患者是否应该接受抗BCG 治疗的问题。然而播散性卡介苗和其他分枝杆菌感染尚未在APDS 中报道。因此,虽然个别患者已接受局部卡介苗感染治疗,但APDS 患者并未常规接受抗分枝杆菌预防。
6.2 免疫球蛋白替代
多数APDS 患者存在抗体缺乏的反复呼吸道感染,曾被误诊为变异型免疫缺陷病和高IgM 综合征等,并曾应用IRT 治疗。在接受长期IRT 治疗的APDS 患者中,笔者观察到约一半以上患者的细菌感染情况得到缓解[2,23]。IRT 通常在无支气管扩张的抗体缺乏患者中以0.4 g/(kg·月)的剂量开始,而在支气管扩张患者中,IRT 以高剂量0.6 g/(kg·月)应用[31]。尽管IRT 在治疗部分APDS 患者时能够有效减少呼吸道感染症状,但IRT 似乎不能阻止疱疹病毒感染、淋巴组织增生、自身免疫性或炎症性并发症以及淋巴瘤的进展[32]。此外,在个别APDS患者中,尽管应用IRT 治疗疗效佳,但支气管扩张仍呈进行性发展。
6.3 免疫抑制剂
自身免疫性血细胞减少对皮质类固醇、利妥昔单抗和脾切除均可有效。利妥昔单抗因治疗复杂的、持续的B 淋巴细胞减少而被人所知[23]。非肿瘤性淋巴组织增生好发于APDS,包括淋巴结病、肝脾肿大、呼吸道和消化道局灶性淋巴组织增生。其中有部分非肿瘤性淋巴组织增生的APDS1 患者在应用利妥昔单抗治疗后均得到较好的疗效[23]。此外,ELGIZOULI 等[33]描述在APDS1 患者中炎症性肠病对强的松和维持剂量的美沙拉嗪具有很好的临床反应。少数患者有发育不良或身材矮小表现,然而有文献报道称,不建议应用生长激素治疗,可能与生长激素使生长激素受体激活,进而激活PI3K 信号通路有关[34]。
6.4 雷帕霉素
雷帕霉素是mTORC 抑制剂,能够使激活的PI3K 信号通路活化水平降低,进而使下游S6K 和4E-BP 产生减少[35]。在ESID-APDS 登记队列中[36],评估26 例患者(1 例患者不包括在内,因为治疗是在诊断APDS 之前开始和终止的,且对治疗的反应没有很好的记录),其中包括APDS1 17 例,APDS2 9 例。治疗的主要适应症是淋巴组织增生、结肠炎和/或细胞减少。在平均随访的1.6年后评估每种症状的药物反应,其中淋巴组织增生对药物的反应性好,而肠道炎症和细胞减少的反应性较差。在应用雷帕霉素治疗的同时服用类固醇激素的8 例患者中,7 例能够停止类固醇激素治疗,1 例能够减少药物剂量。个别患者出现药物的严重毒副作用、淋巴瘤以及少数患者表现为治疗无效,部分患者因不同原因而终止治疗。有趣的是,在治疗3~6 个月后对所有被监测者进行评估均未出现相关的疾病变化,但在较长时间的治疗观察(约为2年)后,多数患者病情得到改善,然而,个别患者出现不同程度的病情恶化。迄今为止的研究虽然支持雷帕霉素治疗APDS 相关的非肿瘤性淋巴增生,但长期应用雷帕霉素的益处和风险仍有待确定。
6.5 PI3Kδ抑制剂
Leniolisib(CDZ173)是一种有效抑制PI3Kδ 催化亚基P110δ 活性的口服制剂,能降低T 细胞内PIP3 磷酸化水平,以及降低AKT 和S6 的磷酸化。目前通过Novartis(NCT02435173)正在研究其对APDS 的治疗。RAO 等[37]描述为期12 周,采用非盲法、多点的临床试验,其中包括6 例APDS 患者,在CT/MRI 上均表现为淋巴结肿大和脾肿大。最初的筛选期为50 余天,包括免疫抑制或免疫调节治疗的清除阶段。随后每例患者均应用Leniolisib,随着时间延长不断增加剂量(10 mg、30 mg 和70 mg,2 次/d,4 周/剂量),在进行为期12 周的Leniolisib 治疗后,每例患者的淋巴结和脾脏体积均显著减小,分别为13%~65%(平均40%),26%~57%(平均39%)。
GSK2269557(Nemiralisib)由Glaxo Smith Kline(NCT02593539)进行的试验[38]应用于APDS 患者,虽然口服抑制剂可能对淋巴组织增生更加有效,但实验表明有呼吸道感染以及支气管扩张的患者应用吸入性抑制剂可能更加有益。目前GSK2269557临床试验结果还未正式报道。
6.6 HSCT
HSCT 是目前临床实施的能够根治疾病、重建免疫功能的最重要甚至是唯一手段。NADEMI 等[32]报道了11 例患者在7 个儿科中心接受HSCT 治疗,移植年龄5~23 岁。其中5 例患者接受了全相合的非血缘供体干细胞,4 例为全相合的同胞供体,2 例为半相合的非血缘供体。术后除3 例患者外其余均停止免疫抑制剂和免疫球蛋白替代治疗。术后随访8 个月至16年,有9 例患者(81%)存活。尽管部分患者在移植后可显著改善临床症状,但其严重的并发症及较高的病死率不容忽视。
7 总结和展望
近年来,随着靶向药物的出现及造血干细胞移植治疗提高了患者的生存率,改善生存质量,但从整体而言,其较高的病死率及严重的并发症仍居高不下。因此,本质研究APDS 显得尤为重要。研究发现关于携带PIK3CD基因突变的父母可以完全不发病,而子代却发病,是什么原因抵消该基因突变的后果,目前仍不完全明了。作为根治性手段的基因疗法尚处于实验研究阶段,相信随着更深层次的研究,将阐明基因突变与疾病发生的因果关联,为基因治疗带来必要的理论依据,从而提高疗效并改善预后。
