儿童溶血尿毒综合征4例报道并文献回顾
2021-04-14肖书娜夏治程颖李勇石步云汤文朱松柏秦晨光许慧黄成姣
肖书娜 夏治 程颖 李勇 石步云 汤文 朱松柏 秦晨光 许慧 黄成姣
湖北省妇幼保健院儿童重症医学科,武汉 430000
溶血尿毒综合征(hemolytic uremic syndrome,HUS)是由多种病因引起血管内溶血的微血管病,属于血栓性微血管病(thrombotic microangiopathy,TMA)的一种,临床以微血管性溶血性贫血(microangiopathic hemolytic anemia,MAHA)、血小板减少和急性肾衰竭为主要表现[1]。临床可分为两种类型即感染相关的 HUS(又称典型HUS)和非典型HUS(atypical HUS,aHUS),后者指家族性或特发性补体替代途径调节异常所致HUS。据估计,每年每百万成年人中有2人和每百万儿童中有330人发展为HUS[2]。aHUS更罕见,发病率为五十万分之一。不同分型,临床预后不同。典型HUS病死率低于5%,但20%~30%可伴有不同程度肾功能不全。aHUS预后较差,超过50%的患者会进展至终末期肾病(end-stage renal disease,ESRD),死亡率高达25%[3]。本文对我院4例HUS患儿进行分析总结,为临床诊疗提供参考。
病例资料
一、一般资料
病例纳入2017年5月至2020年4月4例溶血尿毒综合征患儿,均符合诊断标准。年龄7个月~10岁,年龄范围(4.4±4.8)岁,均为男性,1例典型HUS,3例aHUS,其中一例aHUS患儿(例4)其母因妊娠诱发HUS,进展为尿毒症,持续透析治疗,余3例均无家族史。(表1)

表1 患儿一般情况、主要症状及辅助检查
二、临床特殊表现
本组4例患儿,前驱症状包括发热(3例)、咳嗽(2例)、呕吐(2例)合并中枢神经系统症状(谵妄、抽搐)1例,3例患儿合并高血压。所有患儿均出现黄疸、贫血、深茶色尿,辅助检查提示血小板降低及肾功能损害,存在HUS三联征。尿常规:尿隐血(+++),尿蛋白(++~+++),C3均降低,3例C4降低。血尿素氮峰值为15~34(24.2±7.0)mmol/L,血肌酐峰值为104~432(236.4±131.7)μmol/L(图1),血小板最低值为19~37(27.2±6.6)×109/L(图2),乳酸脱氢酶最高值为1561~2258(1 880.6±339.7)U/L。4例患儿血涂片均可见破碎红细胞,网织红细胞升高,Coombs试验阴性,所有患儿肾脏超声示双肾体积增大、回声增强,例1患儿大便O157阳性,余3例患儿大便培养,O157均为阴性。血氨基酸串联质谱、尿有机酸结果正常。自身免疫性抗体筛查(抗核抗体、抗核抗体谱、抗中性粒细胞胞浆抗体等)均为阴性。2例患儿入院后出现少尿,液体超载;2例患儿尿量尚正常,其中1例患儿出院10个月后患儿再次入院。4例患儿家属均拒绝行肾脏穿刺活检术。
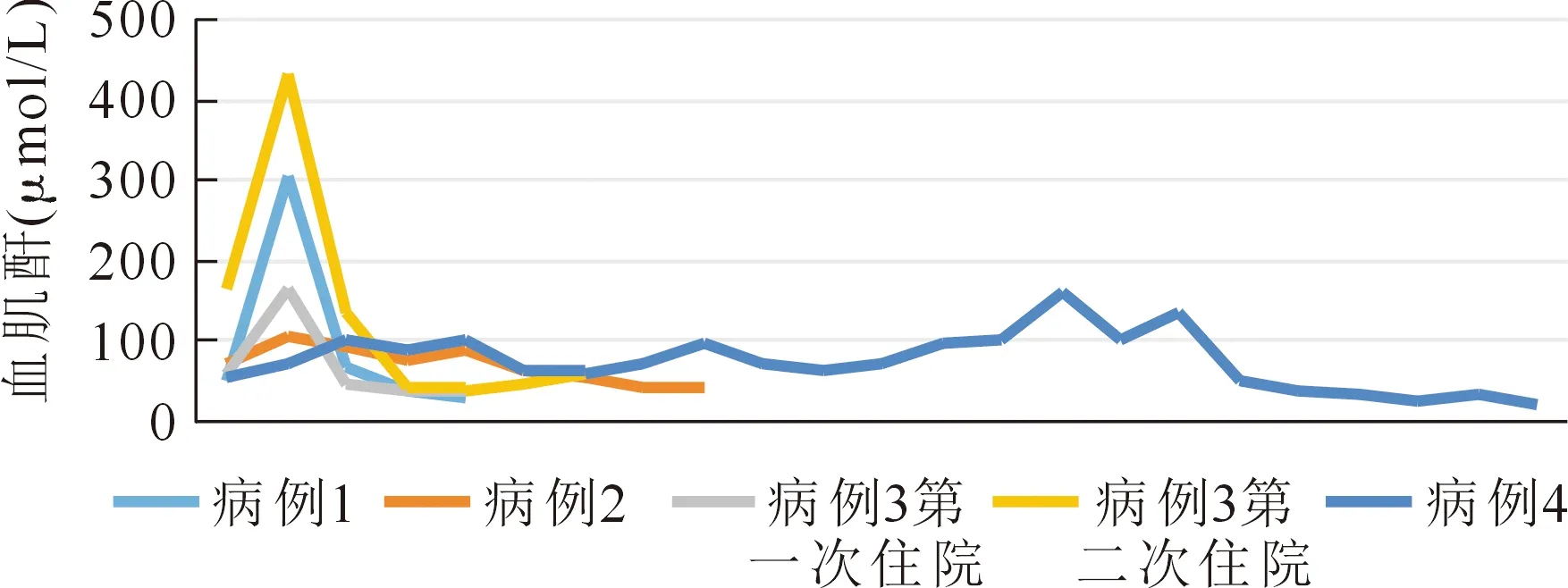
图1 各病例血肌酐变化
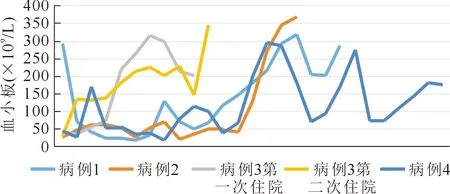
图2 各病例血小板变化
三、治疗及转归
4例患儿诊断明确后立即启动血浆转换(plasma ex change,PE)治疗,其中3例因少尿、肺水肿进行连续性血液透析滤过/血液透析治疗。所有患儿均予以低分子肝素(75~100 U·kg-1·次-1,Q12h),尿激酶(4000~1万U/次,Q8h),甲泼尼龙(12~20 mg·kg-1·d-1),输注洗涤红细胞,输注血小板、血浆、冷沉淀、降压等治疗。3例患儿机械通气。
2例患儿好转,1例复发,1例患儿死亡。例4 患儿入院后予PE治疗5次(600 mL/次),因反复出现肺水肿、肺出血,采取间断机械通气17 d,连续性肾脏替代治疗340.16 h(每周1~3次不等),血尿素氮、血肌酐均未恢复(出院时血尿素氮 11.1 mmol/L,血肌酐78 μmol/L),治疗68 d放弃治疗、拔除气管插管2 h后死亡。患儿基因为THBD突变,c-151G>T,杂合突变。(表2)
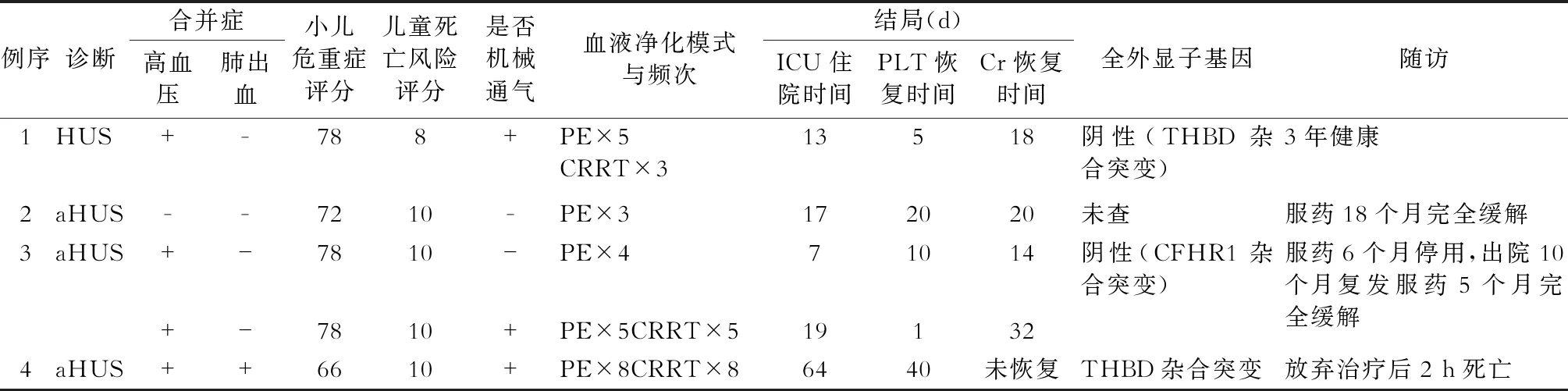
表2 患儿治疗及转归
讨 论
溶血尿毒综合征是小儿急性肾衰竭常见原因之一,典型病例常有前驱胃肠道症状。非典型病例部分有家族史,且易复发,病死率高。近年来,采用血浆置换和透析等综合疗法,病死率明显下降。感染、药物、结缔组织病、器官移植、妊娠、代谢异常等因素可诱发溶血尿毒综合征,典型HUS主要指腹泻相关HUS(Shiga toxin-producing Escherichia coli,STEC-HUS),由产生志贺毒素的肠出血性大肠杆菌(E.coli,EHEC)感染引起细菌性肠炎,其中O157:H7为主要致病原。
例1患儿肠道感染症状起病,入院时肾功能正常,3 d后出现少尿、溶血性贫血、血小板减少、急性肾损伤,大便O157为阳性,诊断为STEC-HUS,患儿全外显子基因检测示THBD杂合突变,c.446G>T,为错义突变,生物学意义为三级,致病性不确定,且基于患儿临床特征,起病急骤、腹泻、血便,经治疗,血涂片第18天未见破碎红细胞,复查C3恢复正常,随访3年患儿身体状况良好,支持STEC-HUS诊断。另3例患儿病初发热或咳嗽,亦为感染起病,但无明显腹痛、便血,例2患儿起病时有皮肤紫癜,住院期间反复鼻出血。3例患儿均具备HUS三联征,肾脏为主要受累器官,乳酸脱氢酶显著升高,C3均不同程度下降,血涂片可见破碎红细胞,网织红细胞升高,尿隐血阳性,Coombs试验均为阴性,骨髓细胞学检查排除恶性血液病等,无服用特殊药物史,诊断为aHUS。同典型HUS一样,以血小板减少、微血管溶血性贫血、肾衰竭为主要表现。临床表现还包括中枢神经系统病变、心力衰竭、呼吸系统疾病、小肠结肠炎、高血压和其他导致多器官多系统功能障碍的状况。本组3例aHUS中,例2未行相关补体因子检测,但例2起病无腹泻,临床进展缓慢;例4有家族遗传史,且血液净化治疗效果欠佳,均诊断为aHUS。
aHUS较HUS病情进展迅速、更易导致多脏器损害且有复发可能,例3合并神经系统症状(谵妄、抽搐),TMA神经系统异常是由于脑微循环血小板血栓形成,最常见意识模糊和精神异常,且呈一过性、反复性或多样性与多变性的特征,头颅CT或MRI检查多无异常发现。该患儿行PE及使用免疫抑制剂有效。但复发后症状较初次加重,且累及心、肺、中枢神经系统等脏器损伤。复发性TMA比初发TMA起病更突然,临床表现更严重,全身受累率更高[4]。例4患儿行PE 6次,血肌酐、血尿素氮持续高于正常,第30天开始连续性肾脏替代治疗/血液透析治疗 1~3次/周,肾功能未逆转,且合并心、肺、胃肠道、凝血功能等多脏器功能损害,需持续透析治疗,PE治疗未能控制病情。
aHUS既可以散发,也可以呈家族聚集性,大约20%病例具有家族遗传背景,与补体基因的特定突变有关。截至2015年,全球共报道了7个补体成分和补体调节基因的先天性遗传异常,即H因子(complement factor H,CFH)、I因子(complement factor I,CFI)、膜辅助蛋白(membrane cofactor protein,CD46/MCP)、C3(component 3)、B因子(complement factor B,CFB)、补体因子H相关蛋白(complement factor H related protein,CFHR)、血栓调节蛋白(thrombomodulin,THBD)和二酰甘油激酶(diacylglycerol kinase ε,DGKE),另外纤溶酶原(plasminogen,PLG)基因突变和INF2(inverted formin 2)也与aHUS发病有关。编码补体调节相关蛋白基因突变导致补体旁路途径过度激活,增加aHUS易感性。THBD是一种内皮糖蛋白,存在于全身所有血管中。在目前报道的aHUS患者中,5%存在THBD编码基因突变,在幼儿中比例更高,提示发病较早。
本组4例患儿,例1患儿全外显子基因检测示存在THBD杂合突变,c.446G>T,为错义突变,生物学意义为三级,致病性不确定,且基于患儿临床特征,起病急骤、腹泻、血便。经治疗,随访3年患儿身体状况良好,支持STEC-HUS诊断。例3患儿血清学检测CFHR显著升高,基因检测示CFHR1杂合缺失,文献报道:意大利154例aHUS患者中,7例(4.5%)存在CFHR基因杂合重排[5]。有学者发现CFHR1缺乏更能诱导产生H因子自身抗体,但其机制尚未明确,该患儿H因子抗体(CFHR)显著升高,同时存在CFHR1杂合缺失,与文献报道一致。例4患儿遗传了其母的THBD杂合突变,反复PE、连续性肾脏替代治疗/血液透析治疗68 d,肾功能未恢复,最终放弃治疗死亡。其母基因分析为THBD相关aHUS,诊断为妊娠相关aHUS(THBD突变),目前肾功能衰竭,尿毒症期,透析治疗。患儿变异染色体为杂合突变,来源于母亲,符合疾病显性遗传模式,位置为chr20.23030292,c.-151G>T,致病性为不确定。目前报道的早发、遗传相关aHUS预后较差。体外研究发现,THBD与CFH和C3b结合,通过加速CFI介导的失活来负调控补体C3b,C3b在辅助因子存在下,从而减弱替代补体级联的激活[6]。THBD是补体系统的负调节剂,THBD的突变可能有助于aHUS的进展。我们对THBD进行体外活性测定,-151G为启动子位点,-151G>T,-151G>A,-151G>C转录水平不会影响基因表达,但蛋白质空间结构异常可能导致蛋白功能异常。因此,需进一步验证此突变位点与aHUS致病性的相关性。文献报道:超过60% aHUS与C3、C4、CFH、CFI、MCP、抗CFH抗体(CFHR)以及CFH、CFI、MCP、CFB和C3突变有关,多为杂合突变且存在不完全外显特点,基因筛查是直接、有效地排查手段。那些具有家族性aHUS病史的孕妇,建议产前行全外显子基因筛查,实现优生优育。
HUS为危及患儿生命的危重症,一旦患儿出现TMA症状时,医师应当开始如下经验性治疗,主要包括控制体液和电解质、控制血压、抗凝、抗感染、纠正贫血以及支持治疗急性肾损伤。洗涤红细胞、血小板输注非HUS常规治疗,本组4例患儿行血液净化治疗前,血小板绝对值均低于20×109/L,且2例合并活动性出血(例2皮肤紫癜、淤血,例4肺出血、消化道出血),予以输注血小板治疗。4例患儿中3例予以输注洗涤红细胞,因其重度贫血及低氧,予以输注洗涤红细胞,提高血红蛋白携氧能力。治疗同时,筛查ADAMTS 13排除TTP,筛查补体相关调节蛋白,一旦确诊aHUS,应在24 h 内开始PE治疗[7]。PE可去除自身抗体和过度活化的补体成分(异常的补体调节蛋白和抗CFH抗体),同时补充正常补体调节蛋白。本组3例aHUS患儿中,例1、例2、例3患儿PE治疗效果显著(见图3、图4,血液净化前、后生化指标对比),例3患儿CFHR升高,行血浆置换可去除血液循环中升高的CFHR,补充正常人的H因子、I因子,减轻补体旁路途径过度活化。例4患儿PE治疗效果欠佳,低龄为其中一个因素,另外推测患儿为THBD基因突变,血浆中无升高的CFHR抗体,仅能置换出过度活化的补体成分。Loirat等[8]总结了THBD相关aHUS经PE治疗的缓解率为60%,40%死亡或进入ESRD,且家族性aHUS患者预后差,ESRD或病死率为50%~80%。本例患儿肾功能不全持续进展,少尿,透析依赖,最终因放弃治疗死亡。因此,婴儿是aHUS患者的一个特殊亚群,疾病进展迅速,死亡率高,在PE治疗病情缓解不明显时,应尽早寻找其他可行有效的治疗方法。
根据2009年的欧洲儿科研究小组的指导方针[9]提出aHUS的一线治疗为依库珠单抗(eculizumab)。患者临床诊断为aHUS后,应考虑使用依库珠单抗治疗[10]。依库珠单抗是一种针对C5的高亲和力单克隆抗体,可阻断C5的裂解,阻断膜攻击复合物MAC的形成,对遗传性和获得性aHUS 患儿均有效,特别适用于PE无效或PE依赖的预后较差的aHUS患儿[11]。有研究表明,对于不同原因导致aHUS,伴或不伴基因突变或自身抗体,依库珠单抗可通过C5阻断末端补体激活以改善补体过度活化,患者均可获益[12]。2011年依库珠单抗已在美国和欧盟地区批准用于aHUS 的治疗[13]。但因其费用昂贵使用受到限制,中国大陆地区尚未临床推广。本组例4患儿,若早期应用依库珠单抗,可能有助于延缓肾功能进展。
综上所述,儿童HUS起病急骤,尤其是aHUS病情凶险,临床表现多样,尽早行PE疗效显著。但对于低龄、有家族史、补体基因异常的患儿,PE治疗可能无效。提倡尽早采用有效治疗方法如依库珠单抗以减少并发症和降低死亡率,但仍需大型临床试验进一步验证疗效及安全性。对于有家族史的患儿,建议其母孕期行产前筛查,指导优生优育。
利益冲突所有作者均声明不存在利益冲突
