烷氧基取代酞菁化合物的合成、表征*
2021-04-11赵美王月霞
赵美,王月霞
(1.榆林职业技术学院 化学工程系,陕西 榆林719000;2.陕西延长中煤榆林能源化工有限公司 质量监督检验中心,陕西 榆林718500)
前言
酞菁化合物可作为非线性光学材料、光限制配合物材料、液晶显示材料、催化剂、分子磁体、光动力学癌症治疗药物、分子电子元器件等,因此对酞菁类化合物的研究具有十分重要的意义。酞菁是由四个异吲哚单元组成的平面大环共轭体系,式1显示了酞菁的分子结构。酞菁(Pc)大环周围的1,4,8,11,15,18,22,25位置被称为非周边位置(np-site)或α位;2,3,9,10,16,17,23,24位置被称为周边位置(p-site)或β位,取代基的位置、种类对酞菁的物理和化学性质影响非常大。通过在酞菁的周边、非周边位置上引入不同的取代基,并对所得到化合物进行比较研究,发现不同的取代基将对酞菁的各种光谱性质[1,2]造成一定的影响,使酞菁配合物具有作为各种分子功能材料的巨大应用潜力。本文介绍了一系列烷氧基取代的自由酞菁、金属酞菁的合成方法和表征方法,获得的各种光谱及数据对研究酞菁化合物有着极大的作用。

式1 Pc的结构
1 实验部分
1.1 试剂
1.1.1 合成M[Pc(β-OC4H9)8](M=2H,Zn)的试剂
邻苯二酚、液溴、DMF、正戊醇(加金属钠蒸馏)、柱色谱硅胶(200~300目)、甲苯、氯仿、二氯甲烷、甲醇、乙醇、五氧化二磷、氨水、石油醚、1-溴代正丁烷、无水K2CO3、无水Na2SO4、氰化亚铜、冰醋酸、金属锂、活性炭、Zn(CH3COO)2·2H2O,所用试剂均为分析纯或化学纯。
1.1.2 合成M[Pc(α-OC4H9)8](M=2H,Zn,Cd,Mn)的试剂
2,3-二氰基-1,4-二氢醌、丙酮、无水K2CO3、碘代正丁烷、甲醇、金属锂、正丁醇、冰乙酸、甲苯、Zn(CH3COO)2·2H2O、C4H6MnO4·4H2O、CdCl2·5/2H2O、DMF、氯仿,所用试剂均为分析纯或化学纯。
1.1.3 合成M[Pc(α-OC5H11)4](M=2H,In)的试剂
正己烷、DMF、乙醇、无水K2CO3、无水MgSO4、无水乙醚、甲苯、金属锂、正戊醇、甲醇、氯仿、InCl3·4H2O,所用试剂均为分析纯或化学纯。
1.2 仪器
Hitachi U-4100型紫外可见分光光度计(测试波长范围300nm~900nm),Bruker DPX 400型核磁共振光谱仪(CDCl3作为溶剂),BIORADFTS-16型红外光谱仪(KBr压片,以2cm-1的分辨率记录),Bruker BIFLEX III型质谱仪(Dithranol为基质),Vario EL III型元素分析仪(以He为载气进行C,H,N测量),JEM-100CXⅡ型透射电子显微镜,Bruker SMART APEXII CCD diffractometer型X-射线单晶衍射仪。
1.3 合成[3,4]
1.3.1 合成M[Pc(β-OC4H9)8](M=2H,Zn)
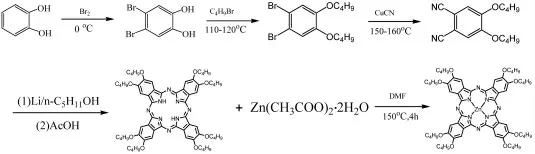
式2 H2[Pc(β-OC4H9)8]and Zn[Pc(β-OC4H9)8]的合成路线
1.3.1.1 合成2,3,9,10,16,17,23,24-八丁氧基自由酞菁
(1)合成4,5-二溴-邻二苯酚
向90mL冷的冰醋酸中溶入20g(0.18mol)邻苯二酚,再把20mL(0.388mol)液溴溶于90mL冰醋酸中,将该溶液滴加到邻苯二酚的醋酸溶液中。缓慢滴加,保证反应完全。滴加完毕,水浴加热下减压蒸馏除去反应中产生的溴化氢和反应混合物中的乙酸。残渣倒入700g冰水中,得到大量灰白色沉淀。过滤,干燥,收率大约85%。
(2)合成4,5-二丁氧基-邻二溴苯
6.7g(0.025mol)4,5-二溴邻苯二酚,0.05mol 1-溴代正丁烷,20g无水碳酸钾在30mL新蒸的N,N-二甲基甲酰胺中,温度控制在110~120℃,搅拌反应8h。冷却得到棕色混合物,将其倒入300mL冷水中,用甲苯萃取,用无水硫酸钠进行干燥。再利用活性碳进行脱色,蒸发甲苯后得棕色粗产品。用乙醇进行重结晶,得9.48g白色晶体,收率为90%。
(3)合成4,5-二丁氧基-邻二氰基苯
9.48g(25.0mmol)4,5-二丁氧基-邻二溴苯,5.3g(59.2mmol)氰化亚铜,62mL N,N-二甲基甲酰胺,温度控制在150~160℃,搅拌反应6h。混合物冷却后,倒入1000mL浓氨水中,通入空气24h。反应混合物变为蓝色时,用800mL甲苯进行萃取,再用水洗涤至pH值为中性,用无水硫酸钠进行干燥。蒸发掉溶剂,用氯仿/石油醚=2∶1作为淋洗液进行色谱柱层析(200~300目硅胶),再用乙醇进行重结晶,得0.96g白色沉淀,收率为14%。
(4)合成2,3,9,10,16,17,23,24-八丁氧基取代自由酞菁
向5mL正戊醇中加入1.95mmol 4,5-二丁氧基-邻二氰基苯和4mmol金属Li,N2保护下加热到120℃反应2h。冷却到室温,再加入100mL甲醇,边搅拌边滴加2mL乙酸,过滤,收集析出的固体,用甲醇洗涤,干燥。再用氯仿为淋洗液在硅胶柱上提纯,收集绿色带,蒸去溶剂,用甲醇/氯仿混合溶剂重结晶。得到0.12g深绿色粉末状固体,收率约5.6%。
1.3.1.2 合成2,3,9,10,16,17,23,24-八丁氧基取代锌酞菁
向4mLDMF中加入0.10mmolZn(CH3COO)2·2H2O和0.05mmol自由酞菁,在N2保护下加热到150℃,回流约4h,减压蒸馏,蒸去溶剂,用氯仿为淋洗液在硅胶柱上提纯,再用甲醇/氯仿混合溶剂重结晶,得到45mg墨绿色针状晶体,收率约为78%。
1.3.2 合成M[Pc(α-OC4H9)8](M=2H,Zn,Cd,Mn)
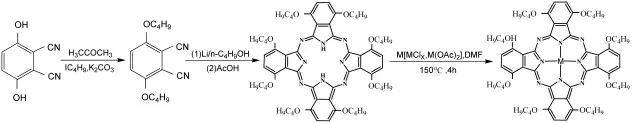
式3 H2[Pc(α-OC4H9)8]and M[Pc(α-OC4H9)8](M=Zn,Cd,Mn)的合成路线
1.3.2.1 合成1,4,8,11,15,18,22,25-八烷氧基取代酞菁(1)合成3,6-二丁氧基-1,2-二氰基苯
N2保护下,1.60g 2,3-二氰基-1,4-二氢醌和150mL干燥的丙酮加入到装有磁子的反应器中,在不断搅拌下加入6.0g碳酸钾和9.2g碘代正丁烷,回流反应60h。冷却后,将所得混合物倒入300mL水中,产品析出,抽滤。得到的粗产品用少量氯仿溶解,加入甲醇重结晶。再次抽滤后,在空气中放置1d,真空干燥1h,得到1.05g白色固体,收率38%。
(2)合成1,4,8,11,15,18,22,25-八丁氧基取代自由酞菁
合成方法与2,3,9,10,16,17,23,24-八丁氧基取代自由酞菁的合成方法类似,见上文。最终得到0.14g深绿色粉末状固体,收率约6.4%。
1.3.2.2 合成1,4,8,11,15,1,22,25-八烷氧基取代金属酞菁
合成1,4,8,11,15,18,22,25-八烷氧基取代酞菁锰、酞菁锌、酞菁镉的步骤与合成2,3,9,10,16,17,23,24-八丁氧基取代锌酞菁类似,见上文。只是加入金属锰盐C4H6MnO4·4H2O、金属锌盐Zn(CH3COO)2·2H2O和金属镉盐CdCl2·5/2H2O,在此不再详细叙述,收率大约分别为10%、20%、9%。
1.3.3 合成M[Pc(α-OC5H11)4](M=2H,In
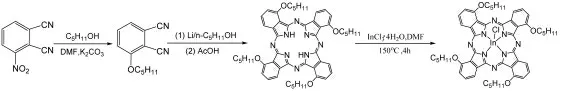
式4 H2[Pc(α-OC5H11)4]and In[Pc(α-OC5H11)4]Cl的合成路线
1.3.3.1 合成1,8,15,22-四烷氧基取代自由酞菁(1)合成3-戊氧基邻二氰基苯
将1g 3-硝基邻二氰基苯、2.5mL正戊醇、8g无水碳酸钾加入到12mL干燥的DMF中,室温下搅拌反应48h。反应混合物倒入100mL水中,用乙醚(100mL*3)萃取,用无水硫酸镁干燥,蒸去乙醚后用硅胶柱提纯,淋洗液为甲苯。蒸去甲苯后真空干燥,得到0.3g产物,收率约24%。
(2)合成1,8,15,22-四烷氧基取代酞菁[5]
将240mg(0.89mmol)3-戊氧基邻二氰基苯和28mg(4mmol)金属Li加入到6mL正戊醇中,在N2保护下加热到110℃,反应4h。冷却到室温后加入100mL甲醇,边搅拌边滴加2mL乙酸,过滤收集析出的固体,用甲醇洗涤,干燥。再以氯仿为淋洗液在硅胶柱上提纯,收集第二个绿色带,蒸去溶剂,用甲醇/氯仿混合溶剂重结晶。得到0.16g深绿色粉末状固体,收率约21%。
1.3.3.2 合成1,8,15,22-四戊氧基取代酞菁铟
合成方法与合成2,3,9,10,16,17,23,24-八丁氧基取代锌酞菁类似,见上文。只是加入金属铟盐InCl3·4H2O(0.10mmol),最终得到墨绿色针状晶体36.5mg,收率约72%。
2 结果与讨论
2.1 电子吸收光谱[6,7,8](UV-Vis)
该系列烷氧基取代酞菁配合物的电子吸收光谱数据均在CHCl3中测得,总结归纳在表1中。由表1可以看出,该系列酞菁配合物在可见光区域(600~800nm)有很强的吸收带(通常称为Q带)。此外,在紫外区(340nm左右)有一个较强吸收带,一般称为Soret带,也称为B带。Q带和Soret带分别对应于酞菁分子能级图中的不同能级间的电子跃迁。
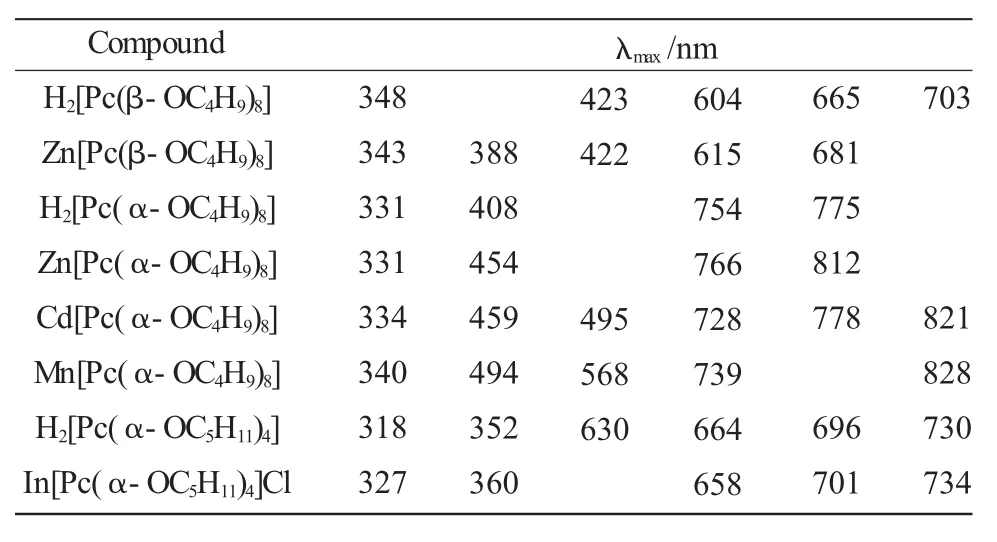
表1 取代酞菁化合物的紫外可见吸收光谱数据Table 1 The UV-Vis absorption spectrum data of the substituent phthalocyanines
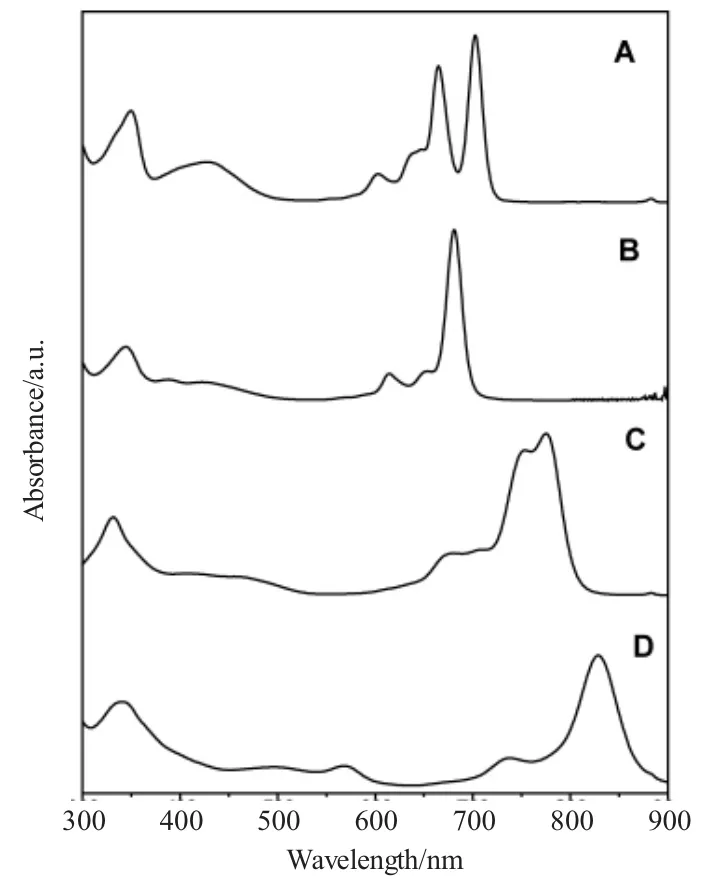
图1 H2[Pc(β-OC4H9)8](A),Zn[Pc(β-OC4H9)8](B),H2[Pc(α-OC4H9)8](C),Mn[Pc(α-OC4H9)8(D)的紫外可见吸收光谱图Fig.1 The UV-Vis absorption spectra of H2[Pc(β-OC4H9)8](A),Zn[Pc(β-OC4H9)8](B),H2[Pc(α-OC4H9)8](C)and Mn[Pc(α-OC4H9)8(D)
根据以上谱图我们可以得出这样的结论,α位取代配合物和β位取代配合物相比,因为酞菁配体非周边位置的取代基比周边位置对酞菁环的影响更大,所以在供电子基团的作用下,可以清楚地看出α位取代配合物的Q吸收带较β位取代配合物的Q吸收带发生了明显红移。
2.2 核磁共振氢谱1H NMR
通过核磁共振测试[9,10],证明分子结构正确无误,且产品的纯度相当高。图2给出了Cd[Pc(α-OC4H9)8]的核磁共振谱图。以图2为例,δ7.44~7.54的双重峰是Pc环上芳香H的信号,δ4.80~4.96的三重峰是OCH2质子的信号,δ1.62~2.14的多重峰是烷氧基链上其它亚甲基H的信号,δ1.03~1.05的三重峰是CH3质子的信号。

图2 Cd[Pc(α-OC4H9)8]的核磁共振氢谱图Fig.2 The 1H NMR spectrum of Cd[Pc(α-OC4H9)8]
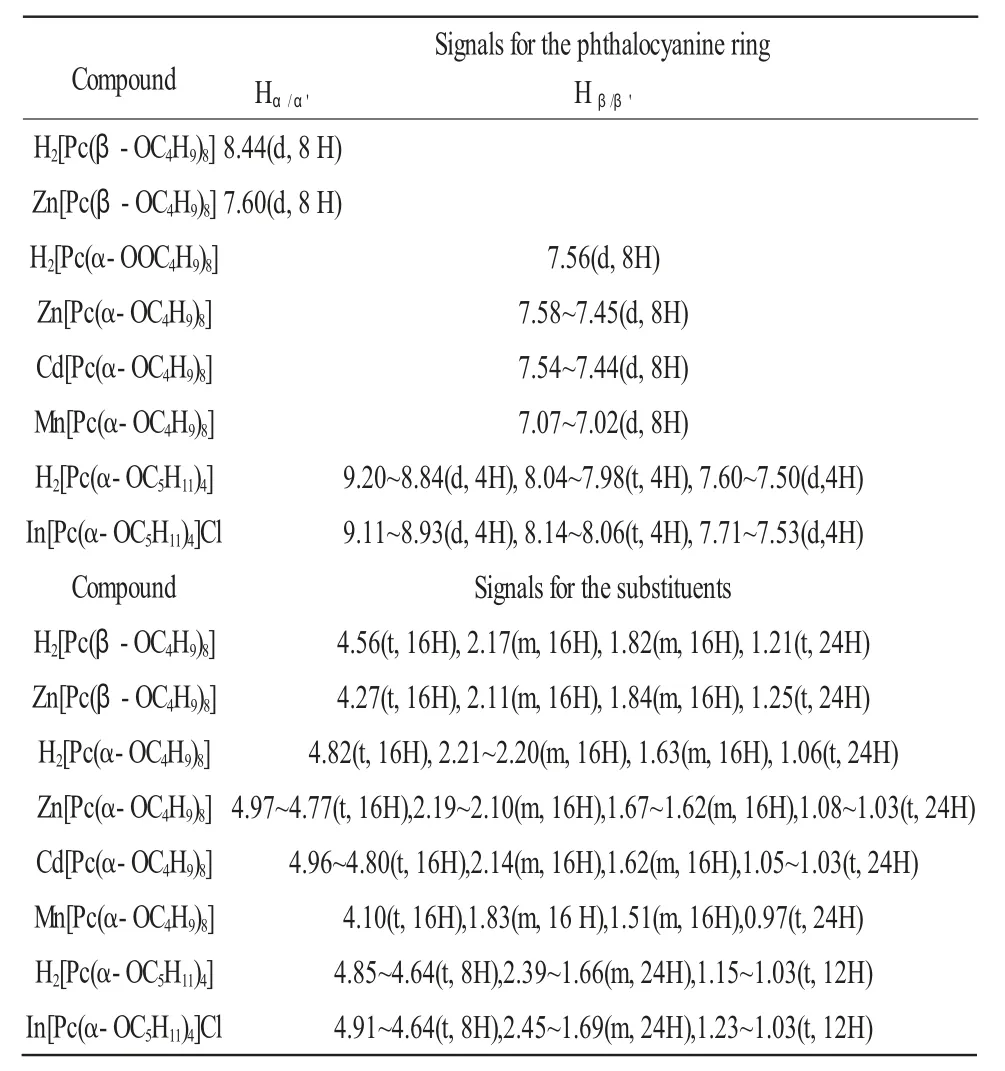
表2 取代酞菁化合物的核磁共振氢谱数据Table 2 The 1H NMR data of the substituent phthalocyanines
2.3 红外光谱IR
运用红外光谱对所合成的一系列酞菁化合物进行了表征,并对所观测到的谱峰进行了指认[11,12,13]。结果显示共有的吸收有系列化合物的异吲哚环伸缩峰、吡咯C=C伸缩峰;H2[Pc(β-OC4H9)8]在1098.85cm-1处,Zn[Pc(β-OC4H9)8]在1095.40cm-1处,Zn[Pc(α-OC4H9)8]在1098.04cm-1处,Cd[Pc(α-OC4H9)8]在1098.75cm-1处,H2[Pc(α-OC5H11)4]在1110.54cm-1处,In[Pc(α-OC5H11)4]Cl在1080.97cm-1处的C-H弯曲峰;H2[Pc(β-OC4H9)8]在743.06cm-1处,Zn[Pc(β-OC4H9)8]在741.74cm-1处,H2[Pc(α-OC4H9)8]在746.40~720.99cm-1处,Mn[Pc(α-OC4H9)8]在740.82cm-1处,H2[Pc(α-OC5H11)4]在746.79cm-1处,In[Pc(α-OC5H11)4]Cl在739.32cm-1处的C-H摇摆峰。

图3 H2[Pc(α-OC4H9)8](A),Zn[Pc(α-OC4H9)8](B),Cd[Pc(α-OC4H9)8](C),Mn[Pc(α-OC4H9)8](D)的红外光谱图Fig.3 The infrared spectra of H2[Pc(α-OC4H9)8](A),Zn[Pc(α-OC4H9)8](B),Cd[Pc(α-OC4H9)8](C)and Mn[Pc(α-OC4H9)8](D)
该系列酞菁化合物独特的吸收有:位于2958~2954cm-1、2932~2922cm-1、2871~2854cm-1处的脂肪侧链C-H对称伸缩振动和不对称伸缩振动峰;另外值得注意的是,对于自由酞菁H2[Pc(β-OC4H9)8]、H2[Pc(α-OC4H9)8]、H2[Pc(α-OC5H11)4]预期中应当出现在高于3000cm-1位置的N-H伸缩振动,由于被相同位置的强烈H2O杂质峰掩盖而无法辨认。
2.4 MALDI-TOF质谱

表3 取代酞菁化合物的质谱数据Table 3 The mass spectrometric data of the substituent phthalocyanines
MALDI-TOF质谱可以给出鲜明的分子离子峰,分子离子峰[M]+或[MH]+的m/z值一般等于产物相对分子量或相对分子量加一,可确切指示特定分子的存在。该系列烷氧基取代酞菁化合物均通过了质谱测试,所测得m/z值均与理论相对分子量相符。表3中给出了各自的质谱数据。
2.5 元素分析EA
为证实所合成的一系列烷氧基取代酞菁化合物的分子组成以及证明其纯度,对所有化合物均进行了元素分析(C、H、N)表征。在重复柱分离和重结晶之后,新合成的这些酞菁配合物都得到了满意的元素分析数据,见表4。
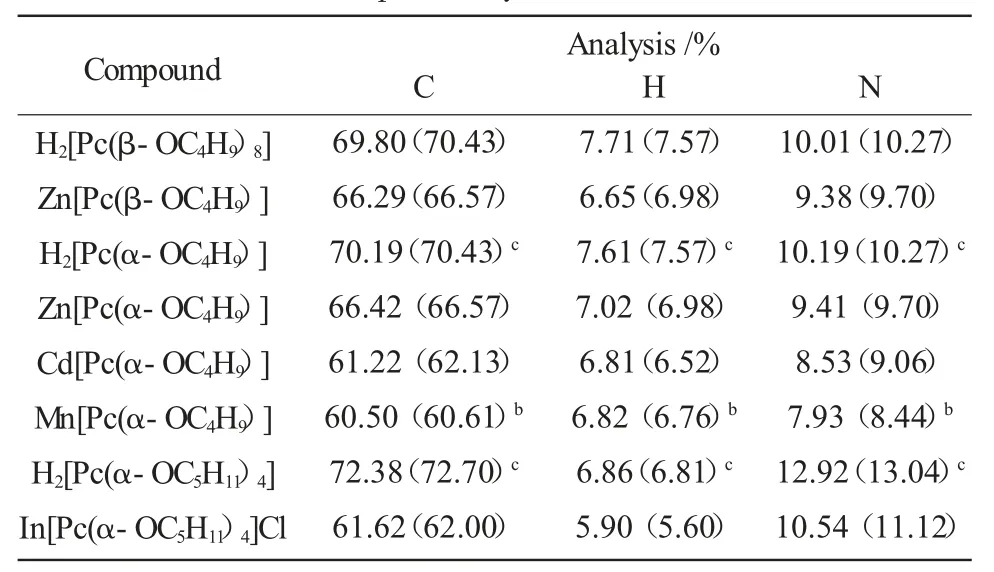
表4 取代酞菁化合物的元素分析数据Table 4 The elemental analysis data of the substituent phthalocyanines
2.6 化合物的自组装形貌

图4 H2[Pc(β-OC4H9)8]的TEM图像Fig.4 The TEM images of the H2[Pc(β-OC4H9)8]

图5 H2[Pc(α-OC5H11)4]的TEM图像Fig.5 The TEM images of the H2[Pc(α-OC5H11)4]
自组装可以创造具有新颖结构和功能的有序分子聚集体,具体是通过分子间相互作用形成的[14]。首先配制10-3~10-5mol-1的样品溶液,溶剂选用氯仿,在另一个小玻璃瓶中加入一定量的甲醇溶液,然后用微量注射器吸取30~50μL的样品溶液,快速注入到甲醇溶液中,放置30min左右,用滴管吸取少量组装之后的溶液滴加在覆盖有碳膜的铜网上,就可以用于TEM的测试了。图4中的A图是放大50000倍后得到的图像,B图是放大14000倍得到的图像,从图中可以看出H2[Pc(β-OC4H9)8]自组装后得到的是纳米棒状结构;图5中的A,B图均为放大10000倍后得到的图像,可以看出H2[Pc(α-OC5H11)4]自组装后得到的是纳米线状结构。
2.7 化合物的结构研究
单晶的培养是通过溶剂挥发法来实现的,甲醇慢慢扩散到盛有Zn[Pc(α-OC4H9)8]氯仿溶液的小玻璃瓶中,大约20d左右生成了适合X-射线分析的晶体。图6是Zn[Pc(α-OC4H9)8]的晶体结构图,表5列出了Zn[Pc(α-OC4H9)8]的晶胞参数和部分测量参数。这个晶体结构属于单斜晶系,四个这样的配合物分子存在于同一个晶胞中,这四个分子是面对面交错着排列在一起的。
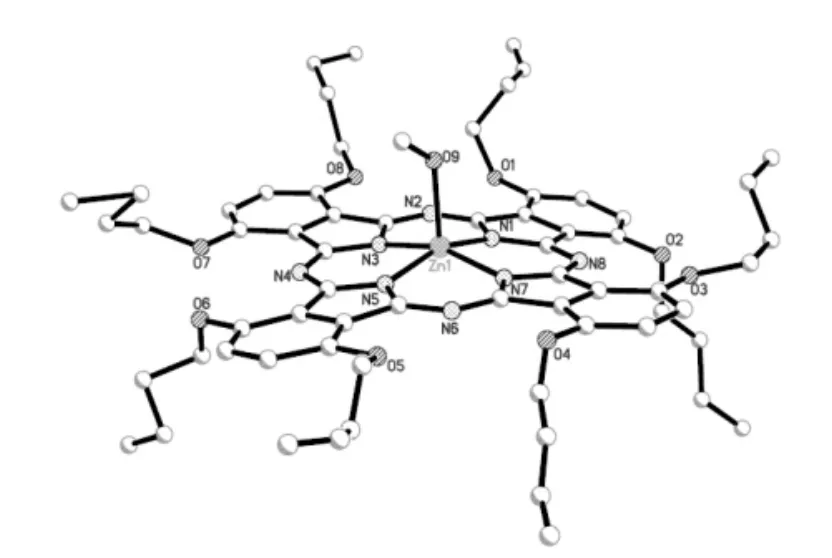
图6 Zn[Pc(α-OC4H9)8]的晶体结构图Fig.6 The crystal structure diagram of the Zn[Pc(α-OC4H9)8]
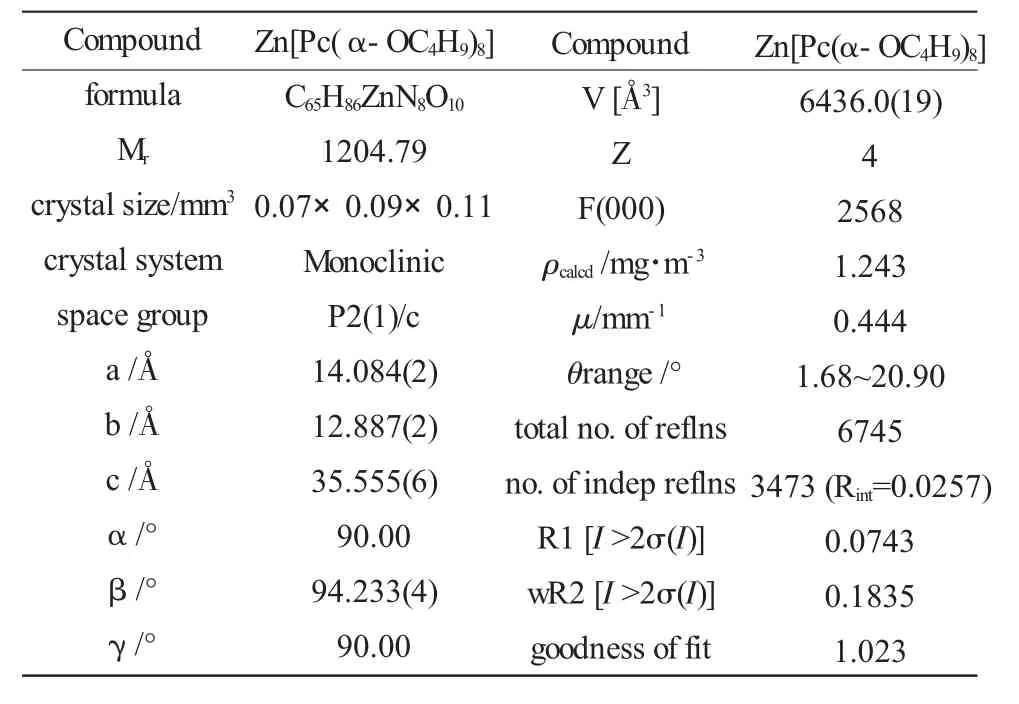
表5 Zn[Pc(α-OC4H9)8]的晶体结构数据Table 5 The crystallographic data of the Zn[Pc(α-OC4H9)8]
3 结论
我们合成了一系列含有不同烷氧基取代的酞菁化合物[15],并利用各种光谱对这些化合物进行了充分的表征。结果表明,酞菁配体上取代基的种类、数量和位置对化合物的各种光谱性质均有一定的影响,从而为合成新型酞菁并作为特殊的功能材料奠定了一定的基础。
此外,通过对一系列化合物在甲醇溶液中进行自组装测试,发现H2[Pc(β-OC4H9)8]、H2[Pc(α-OC5H11)4]能够分别形成纳米棒状结构和纳米线状结构的自组装形貌,说明酞菁化合物作为功能材料有着巨大的应用潜力,可以通过自组装技术获得期望的材料结构。
利用溶剂挥发法得到了Zn[Pc(α-OC4H9)8]的单晶,经过X-射线单晶衍射仪测试后,获得了该化合物的一系列相关晶体数据,为更好地掌握化合物的性质提供了依据。
