烟草烟雾暴露结合基因工程小鼠在COPD药物靶点及机制研究的应用进展
2021-04-09任周新沈俊岭刘海军余海滨李建生
任周新,赵 迪,沈俊岭,刘海军,余海滨,李建生
(1. 河南中医药大学中医药科学院,河南 郑州 450046;2. 呼吸疾病中医药防治省部共建协同创新中心,河南 郑州 450046;3. 河南中医药大学第一附属医院,河南 郑州 450000)
基因工程技术包括ES细胞打靶技术、CRISPR 基因编辑技术、转基因技术等,随着技术的日臻完善,人类改变基因的能力正迅速增强。目前,在改变基因的实验动物中,小鼠的应用频率最高,建立了众多与人类疾病有关的模型[1]。COPD的形成与恶化,决定于内在基因与外在环境因素的共同作用。作为最主要的致病因素烟草烟雾暴露,结合不同的转基因小鼠,能够阐释某种特定基因表达的变化对烟草烟雾暴露动物肺损伤、关键信号通路等的影响,阐释特定基因或信号通路在COPD病理机制中的作用,确定新的药物作用靶点。本文就烟草烟雾暴露结合基因工程小鼠在COPD病理机制和药物靶点方面的研究结果,进行了综合和分析,报导如下。
1 药物靶点
1.1 Tristetraprolin (TTP)TTP是由锌指蛋白36 (zinc finger protein-36,ZFP36) 基因编码的一种RNA结合蛋白,通过与靶细胞因子的3′-非翻译区的富含腺苷酸和尿苷酸的多种元件的结合,诱导mRNA的破坏,特别是某些编码炎症的细胞因子的转录本的破坏[2],可能对COPD具有保护作用。TTP降低mRNA稳定性的作用,被磷酸化和脱磷酸化的两个特定的残基所调控,在小鼠是磷脂酰丝氨酸52和178,在人类是磷脂酰丝氨酸60和186[3]。mitogen-activated protein kinase (MAPK)-activated protein kinase 2 (MK2)磷酸化TTP,并使之失活,导致靶mRNA的稳定[4]。蛋白磷酸酶2A(PP2A),使TTP脱磷酸化,提高其活性,促进靶mRNA的稳定性丢失[5]。而活化的TTP又被泛素-蛋白酶体系统作为靶点,被其降解[5]。这些方式中,关键的磷脂酰丝氨酸的磷酸化控制了TTP的功能,这些磷脂酰丝氨酸如同一个分子开关:磷酸化,功能关闭,脱磷酸化,开放。所以,如果将TTP作为一个新的抗COPD的抗炎靶点,它具有易于控制的优点。Nair等[5]使用了一种基因修饰的组成型的TTP活化小鼠品系,其内源性TTP蛋白的磷脂酰丝氨酸52和178被不能被磷酸化的丙氨酸残基所取代(Zfp36aa/aa)[6]。这两个氨基酸残基的置换,导致TTP结构性的活化,起到一种mRNA脱稳定性的作用。应用Zfp36aa/aa小鼠,检测活化的TTP对实验性烟草烟雾(cigarette smoke,CS)诱导COPD的影响。发现活化的TTP对实验性COPD的多种特征具有保护效应,如肺炎症减轻、气道重构减轻、肺泡扩张减轻和肺功能改善。随后在野生型的C57BL/6J小鼠中,用一种PP2A激动剂(AAL(s)),以活化TTP,单独或合用蛋白酶体抑制剂bortezomib,随后评估这些干预对CS诱导的实验性COPD的影响。结果表明药物活化TTP能够减轻疾病的典型表现。该研究结果展示出应用药物活化TTP,具有减轻COPD的潜能。
1.2 晚期糖基化终末产物受体(receptor for advanced glycation end products, RAGE)晚期糖基化终末产物受体是一种多配体的受体。烟草烟雾中含有RAGE的配体,已经被证实是COPD的病原之一。野生型和RAGE基因敲除小鼠,分别暴露于CS。比较后发现,RAGE基因敲除小鼠的肺组织炎症病变明显减轻,肺泡灌洗液中炎症相关因子和中性粒细胞数量减少。该结果表明敲除RAGE,能缓解CS诱导的气道炎症。cDNA微列阵比较后发现,作用途径可能为S100A8/A9的表达下调以及与之相关的免疫-炎症反应下调[7]。另有研究结果发现[8]:敲除RAGE基因或者应用RAGE抑制剂FPS-ZM1,阻止CS诱导小鼠的肺气肿的进展。对暴露于CS的敲除RAGE小鼠或C57BL/6野生型小鼠的肺泡巨噬细胞执行了无偏基因表达谱分析,通过比较,识别出一些COPD发病机制必需的基因,表明在CS诱导的巨噬细胞活化中,RAGE是必需的组成部分[8]。两种来源的细胞,最显著的差异是Nrf2介导的基因表达。敲除RAGE小鼠巨噬细胞Nrf2介导的基因表达和肺组织4-羟基壬稀醛 (4-hydroxynonenal,4-HNE)减少,说明在CS刺激时,敲除小鼠的肺氧化应激水平较低。但是,两种小鼠CS暴露相同、肺泡灌洗液中的巨噬细胞数量相似、肺组织CD45阳性表达细胞和中性粒细胞阳性表达细胞的百分率也相似[8],提示CS介导的氧化应激可能需要RAGE的介导。另外,对于CS暴露,敲除小鼠巨噬细胞表现出内质网应激的减弱[8]。这些研究表明:在CS诱导的肺巨噬细胞的活化过程和肺损伤过程中,RAGE具有重要的作用;抑制RAGE是COPD治疗的新的可能的途径。
1.3 衰老细胞细胞的衰老由Cdkn2a基因座所介导,其编码两个重要的肿瘤抑制因子,命名为p16Ink4a和 p19Arf [9]。Hashimoto等[10]通过消融Arf表达的细胞,构建了一种ARF-DTR转基因小鼠,提高了小鼠的肺功能。预先用白喉毒素清除Arf表达的细胞,可减轻弹性蛋白酶诱导的肺损伤,对肺组织产生一定的保护效应[11]。进一步的研究发现:长期的烟草烟雾暴露导致肺功能异常,肺形态结构异常,衰老标志性蛋白Ink4a和Arf在肺组织的表达增加;在烟草烟雾暴露前,清除Arf表达的细胞,减轻了上述烟雾诱导的表型变化。另外发现:在肺损伤后,清除Arf表达的细胞,在烟草烟雾提取物攻击的小鼠中,也产生了有益的影响;初步的研究结果发现,清除衰老细胞的保护效应,与巨噬细胞等分泌的弹性裂解酶的产量或活性降低有关[12]。这些结果提示:衰老细胞是COPD重要的治疗靶点。
2 炎症和免疫反应
2.1 正常肺和炎症肺正常的肺和炎症的肺对于CS刺激反应的比较,对于理解COPD的形成与发展是有益的。βENaC小鼠,是一种转基因过表达Scnn1b的小鼠,该基因能够编码存在于气道的上皮细胞的Na+通道β亚单位(βENaC)。βENaC小鼠具有脱水的气道黏液,自发形成慢性支气管炎和肺气肿,包括黏液细胞化生、黏液高分泌、轻度的中性粒细胞性炎症、大的泡沫状巨噬细胞以及气道黏液和气道壁的淋巴细胞数量的增加[13]。Engle等[14]应用野生小鼠和βENaC慢性肺炎小鼠,暴露于CS或对照空气,分别暴露1 d和5 d。在肺组织或肺泡灌洗液中,对照βENaC小鼠的白细胞数量、中性粒细胞数量、巨噬细胞数量和淋巴细胞数量均高于对照或CS野生小鼠,反映了βENaC小鼠自发性存在肺组织炎症。CS刺激后,野生小鼠肺出现轻度的炎症反应。与βENaC对照组比较,CS暴露的βENaC小鼠白细胞数量、巨噬细胞数量和淋巴细胞数量均未见明显变化;但中性粒细胞数量,尽管在暴露1 d,没有差异;但在暴露5 d后,CS暴露的βENaC组反而显著减少。两种模型显示出:暴露1 d的炎症介质的变化较暴露5 d的介质变化,改变的数量更多。转录组学的结果显示:暴露1 d或5 d,野生或βENaC小鼠的许多基因和基因集的反应相似,如氧化应激的相关基因上调而免疫反应的基因下调。仅有少许基因的调节和生物反应存在不同:与对照的空气暴露组比较,烟雾暴露组的细胞外基质相关基因和基因集,在暴露1 d上调,但在5 d下调。综合分析,这些结果表明,一次烟雾暴露后,肺基因表达发生了快速的变化,重复性的短期暴露没有明显改变基因表达的变化;在已经出现炎症的肺组织,通过下调某些炎症反应,对重复的后续烟雾刺激损伤,具有一定的保护作用。该结果支持下述假说:对于烟雾刺激,早期的肺组织快速产生一些反应,对于重复性肺损伤,产生保护作用[14]。该假说从某种程度上阐释了吸烟者需要多年的吸烟史,长期而持续的烟雾刺激,才可能导致气道和肺泡的病变,最终形成COPD。
2.2 继发性免疫反应细胞在COPD中,肺组织出现继发性免疫反应细胞数量的增加,主要是浸润的CD4+T,CD8+T和B淋巴细胞。CD8+T不足的小鼠(CD8-/-),而不是CD4+T不足的小鼠,对CS诱导的肺损伤,具有一定的抵抗性,特别是阻止巨噬细胞的积聚、MMP2和MMP9活化以及肺气肿[15],表明在疾病的病理机制中,CD8+T细胞起到了某种关键的作用。与野生型对照组比较,缺乏成熟的T细胞和B细胞(Rag2-/-)的小鼠,在炎症和肺气肿方面,没有变化[16]。然而,从CS暴露的野生小鼠中过继输送CD3+T细胞到Rag2-/-小鼠,后者就形成了类似COPD的表现,说明在COPD的形成和发展中,病原性的T细胞具有某种关键的作用。长期暴露于CS的野生型小鼠与Rag1-/-小鼠,肺炎和肺功能没有差异,但Rag1-/-小鼠气道胶原沉积更为严重,IL-33,IL-13以及ILC2的数量也增加;说明在实验性的COPD中,T/B淋巴细胞在气道胶原沉积和纤维化中,发挥作用,但在炎症中没有作用[17]。
2.3 2型固有淋巴细胞2型固有淋巴细胞(group 2 innate lymphoid cells,ILC2s),在沟通天然免疫反应和继发性免疫反应以及维持肺内环境稳态方面,发挥重要的作用[18]。ILCs细胞属于组织定居的淋巴细胞,根据不同的刺激,这些细胞显示出可塑性[19]。在COPD中,用IL-1β、IL-4和IL-12等刺激,ILC2s发生表型转化,形成了ILC1s细胞或ILC3S细胞;ILC1S:ILC2S比率的增加,与肺功能降低以及疾病的严重性之间,存在直接的相关性[19]。ILC2不足的转基因Rorafl/flII7rCre小鼠与野生型小鼠,长期给予CS暴露,尽管Rorafl/flII7rCre小鼠的肺气肿程度减轻,但胶原沉积以及IL-33和IL-13的表达没有改善[17]。这些研究提示:ILC2s细胞在实验性COPD中,在CS诱导的气道重构和肺气肿中,产生一定的保护作用,但对炎症没有影响。
2.4 DNER/Notch通路在COPD的临床研究中,全基因组关联(genome-wide association studies,GWAS)方法得到了广泛的应用,成为获取目标性状关联的候选基因或基因区域的有力工具[20]。应用该技术,Busch等[21]发现吸烟者气道上皮的DNER(Delta Notch like epidermal growth factor related receptor,DNER)内在区的一个单核苷酸多态性(SNP)与FEV1/FVC 比率或 FEV1之间,存在最显著的相关性;与COPD的遗传风险之间也存在关联。
DNER是一种跨膜蛋白,属于非经典的Notch配体家族,它的膜外段与Notch1受体结合,以一种独立于CSL(CBF1 Suppressor of Hairless,Lag-1)的方式,活化Notch通路。在人的肺组织中,DNER是促炎巨噬细胞的主要表型[22]。通过应用DNER基因工程小鼠,结合烟草烟雾暴露,进一步阐释了DNER/Notch通路的作用机制:
首先,从烟草烟雾暴露的野生型小鼠中,发现肺组织巨噬细胞DNER表达升高,接着发现,促炎的M1骨髓源性巨噬细胞(M1 bone marrow derived macrophages,M1BMDM)DNER的表达升高[23]。随后的研究选择了一种敲除了DNER的小鼠,没有发现敲除DNER对巨噬细胞的极化有影响,推测DNER在M1BMDM的高表达,可能通过调节信号通路发挥作用[23]。采用基因集富集分析(gene set enrichment analysis,GSEA)比较野生小鼠和敲除DNER小鼠的M1BMDM,发现干扰素(interferon,IFN)信号通路在野生小鼠最为富集,两者差异明显,这可能反映了敲除DNER小鼠的M1BMDM存在IFN信号通路的破坏或不足。接着发现敲除DNER小鼠的M1 BMDM细胞受到刺激后,II型干扰素(type II interferon,IFNγ)诱导表达显著降低;GESA显示M1的IFNγ信号富集,敲除小鼠低而野生小鼠高。尽管T细胞是IFNγ的主要来源,然而已经分化的野生小鼠T细胞亚群表达很低的DNER,提示T细胞的DNER不可能调节产生大量的IFNγ;因此,高表达DNER的M1巨噬细胞才是导致IFNγ高表达的原因[23]。在随后的体内实验中,野生型小鼠和敲除DNER小鼠分别暴露于CS,结果发现:在M1巨噬细胞内,DNER表达被启动;IFNγ增加,需要DNER的表达;在慢性炎症中,DNER调节巨噬细胞分泌IFNγ;产生IFNγ的巨噬细胞属于募集的巨噬细胞而不是定居的巨噬细胞;在肺募集的巨噬细胞内,DNER调节IFNγ的分泌。最后发现敲除DNER小鼠的巨噬细胞内,NICD1的核转位显著减少;两种小鼠巨噬细胞的GSEA比较,野生小鼠的Notch1信号通路更为富集。据此认为:借助于DNER,CS诱导体内肺巨噬细胞的Notch1信号通路的活化,随后诱导IFNγ高表达。
3 总结与展望
目前,COPD的个体化治疗已经成为共识,个体基因的异常是导致COPD出现不同临床表型的重要因素[24]。个体化治疗的本质应该反映COPD基因型在致病条件下的复杂而丰富的差异性变化。
随着分子生物学、生物信息学等技术的发展,人们已经识别出了越来越多的COPD表型及其特异性的基因,建立了一种较为成熟的研究模式(Fig 1)。首先,通过GWAS等技术检测正常人和COPD患者,发现表达差异性强的基因和基因座,作为潜在的COPD易感基因和基因座。然后,选择改变某个待筛查基因的小鼠和作为对照的野生小鼠,分别给予烟草烟雾刺激,比较两者间COPD重要指标的变化;另外,应用激动剂或拮抗剂干预该基因在COPD野生小鼠的表达,再次验证该基因是否与COPD的发生和发展有关。这种模式,在探索COPD发病机制和药物干预靶点的研究中,已经得到了应用,取得了一定的进展。
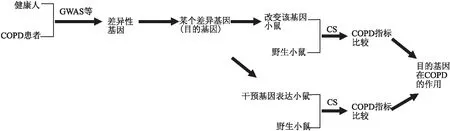
Fig 1 Pattern of combination of cigarette smoke exposure and genetically engineered mice for interest genes searches in COPD
未来,烟草烟雾暴露结合基因工程小鼠的研究模式,将推进COPD复杂表型的准确识别,促进由不同的靶基因改变小鼠结合烟草烟雾暴露的COPD模型体系的建立与完善,推动COPD个体化治疗的建立与发展。
