三例散发I型神经纤维瘤病NF1基因突变检测
2021-04-09马盼盼刘芙蓉郝胜菊张庆华周秉博孙庆梅
马盼盼 张 钏 王 兴 刘芙蓉 郝胜菊 张庆华 陈 雪 周秉博 孙庆梅
甘肃省妇幼保健院,甘肃兰州,730050
I型神经纤维瘤病(neurofibromatosis type1, NF1, OMIM: 162200)是一种常见的常染色体显性遗传病[1]。该病发生于儿童早期,几乎所有的患者都会身体多处出现咖啡色斑,这些色斑随着患者年龄的增长而增多增大。腋下和腹股沟的色斑出现儿童晚期。大部分成年患者会发生皮肤神经纤维瘤,一般是发生于皮肤上或稍下部的良性肿瘤[2]。手术切除丛状神经纤维瘤效果不佳,对于恶性外周神经鞘肿瘤可进行手术切除。除此之外,其临床表现还有骨骼异常、Lisch结节、学习障碍等。其发病率约为1∶4000至1∶3000,其中约50%是自发突变,成人外显率接近100%[3,4]。
I型神经纤维瘤病是由肿瘤抑制基因NF1突变所致。NF1基因位于常染色体17q11.2,由61个外显子组成[5]。NF1基因的表达产物为神经纤维蛋白。作为“肿瘤抑制因子”,神经纤维蛋白通过激活Ras-GTPase而发挥作用,它的表达受损使Ras活性持续增加,可导致细胞增殖[6]。除了调节Ras活性外,神经纤维蛋白还可以正向调节环状单磷酸腺苷(cAMP)的水平。与Knudson的“两次打击”假说一致,NF1基因突变的患者会出现一系列I型神经纤维瘤病症状,例如咖啡斑、神经纤维瘤、胫骨发育异常和视神经通路肿瘤[7]。
刘芷兮等[8]总结了NF1 的突变类型及其基因与表型关系。NF1 基因突变包括无义突变、错义突变、基因大片段缺失与剪切突变和移码突变等,这些突变类型我国均已有相关文献报道。本研究对3例散发患者进行基因检测,现将结果报道如下。
1 资料与方法
1.1 临床资料 3例患者均来自甘肃省妇幼保健院医学遗传中心。患者1,女,6岁,出生后不久右上肢出现水肿,右胸部肿大,躯干部有多个牛奶咖啡斑,并逐渐增多增大。查体可见躯干部皮肤较多咖啡斑,右胸部有明显肿物(图1),近半年头晕症状加重。患者父母均正常,无家族史。患者2,女,18岁,6岁时腋窝、腹部、大腿内侧逐渐出现密集小牛奶咖啡斑。同时伴肾动脉血管狭窄、继发性高血压。查体见皮肤多处色素沉着(牛奶咖啡斑),腋窝、腹部、腹股沟有雀斑。患者母亲自幼眼睑、左侧肘部外侧有较小的色斑;患者兄长及父亲均未见显著皮肤色素沉着,无家族史。患者3,女,6岁,全身皮肤可见较大咖啡斑,同时伴有生长激素缺乏性矮小症(110 cm,血清生长激素0.27 ng/mL)。患者父母均正常,无家族史。
1.2 标本采集 患者1和患者3分别抽取患者及其父母外周血各2~3 mL,患者2抽取患者、患者父母及其兄长外周血各2~3 mL,所有标本均收集于5 mL EDTA抗凝管中,于-80℃保存。3例患者及其家属均签署基因检测知情同意书。
1.3 基因组DNA提取 取200 μL标本,采用全自动核酸提取仪提取所有标本DNA,然后使用核酸浓度测定仪NanoDrop2000进行标本DNA浓度测定。所有标本DNA浓度在50 ng/μL左右,OD260/280在1.8~2.0之间。
1.4 NF1、NF2基因高通量测序及生物信息学分析 样本送检至迈基诺基因检测公司进行高通量测序,平均测序深度为1000×,所有区域达到20×以上的覆盖度。
1.5 Sanger及MLPA法验证 点突变验证分析:根据NF1基因序列(NM_000267),使用在线Primer 3.0(http://primer3.ut.ee)软件设计引物。对候选致病突变位点所在基因组区域进行PCR扩增,纯化PCR扩增产物,使用BigDye测序反应试剂盒(美国Applied Biosysterms公司)进行测序反应,采用ABI 3500D Genetic Analyzer仪器(美国Applied Biosysterms公司)进行检测。测序结果与NF1基因参考序列(NM_000267)进行序列比对。
外显子杂合缺失分析:采用多重探针连接扩增技术 (multiplex ligation dependent probe amplification,MLPA)检测试剂盒(SALSA MLPA P081及P082探针,荷兰MRC公司),实验经过DNA的变性、探针杂交、连接反应、扩增及产物分析可检测NF1基因拷贝数的变化。
1.6 亲缘关系鉴定 使用中德美联亲子鉴定套装1(Expressmarker 22)试剂盒对患者及其父母亲缘关系进行鉴定分析。按照试剂盒说明书完成患者及其父母DNA样本PCR扩增。之后将PCR扩增产物在ABI 3500D Genetic Analyzer测序仪上进行毛细管电泳。应用GeneMapper软件进行电泳结果分析。
2 结果
3例散发I型神经纤维瘤病患者中,共检出NF1基因3个突变(表1)。包括2个杂合缺失和1个无义突变,分布于NF1基因的各个功能区,无明显的分布热区。我们通过亲子鉴定分析3例患者与其父母均符合亲缘关系,Sanger测序和MLPA验证结果显示3例患者父母均不携带这些突变,说明3例患者所检测到的突变均为自发突变,见表1。
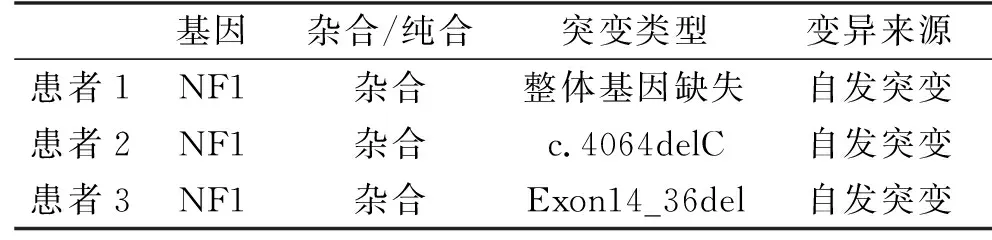
表1 3例患者突变结果图
3例患者中患者1为NF1基因整体杂合缺失突变,HGMD专业版数据库已有文献报道与NF1相关,且为致病性突变[9];用MLPA法进行家系验证分析,其父母该基因均无缺失,此变异为自发突变,见图2。患者2分析到NF1基因有1个杂合突变c.4064delC,该变异为无义突变,导致氨基酸改变p.S1355X(即1355位的丝氨酸发生终止突变),可能导致基因功能丧失;此外经家系验证分析,患者之父母及兄长均未检测到该位点变异(图3),此变异为自发突变;该变异在正常人群数据库中的频率未见统计,为低频变异;根据ACMG指南,此变异初步判定为致病性变异(Pathogenic)。HGMD数据库未有该位点的相关性报道;生物信息学蛋白功能预测软件SIFT(https://sift.bii.a-star.edu.sg/www/SIFT4G_vcf_submit.html)、PolyPhen_2(http://genetics.bwh.harvard.edu/pph2/)、MutationTaster (http://www.mutationtaster.org)、GERP++(http://mendel.stanford.edu/SidowLab/downloads/gerp/index.html)分别预测为未知、未知、未知、未知。患者3检测到NF1基因14-36号外显子杂合缺失,HGMD数据库未有该位点的相关性报道;用MLPA法进行家系验证分析,其父母该基因均无缺失,此变异为自发突变,见图4。
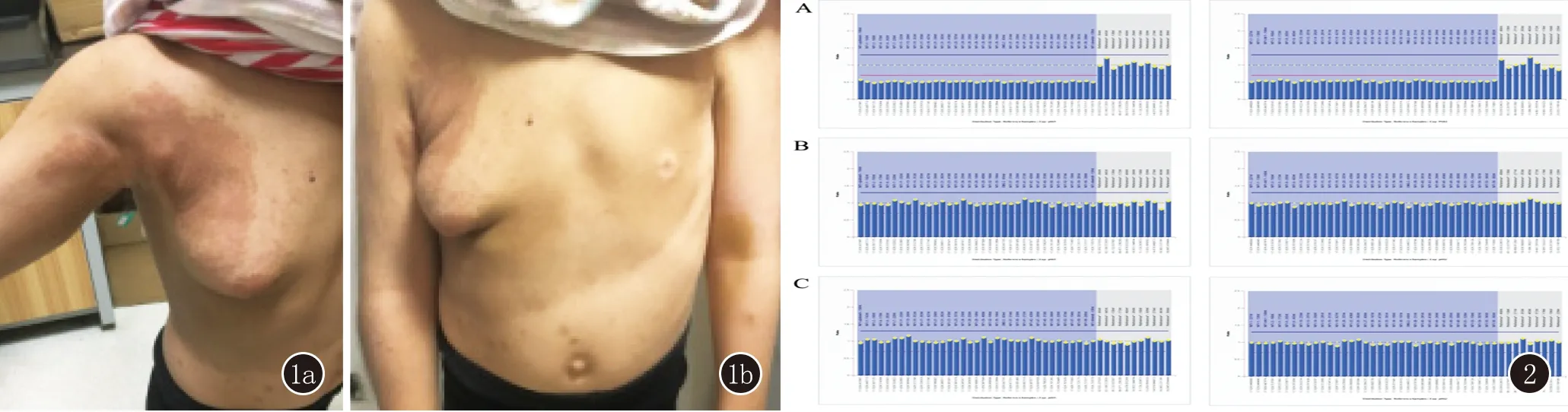
图1 患者1侧面(1a)和正面(1b),躯干部皮肤较多咖啡斑,右胸部有明显肿物 图2 患者1家系突变结果图 A为患者1患者 NF1基因整体杂合缺失;B、C分别为患者之父、之母NF1基因未见相应缺失
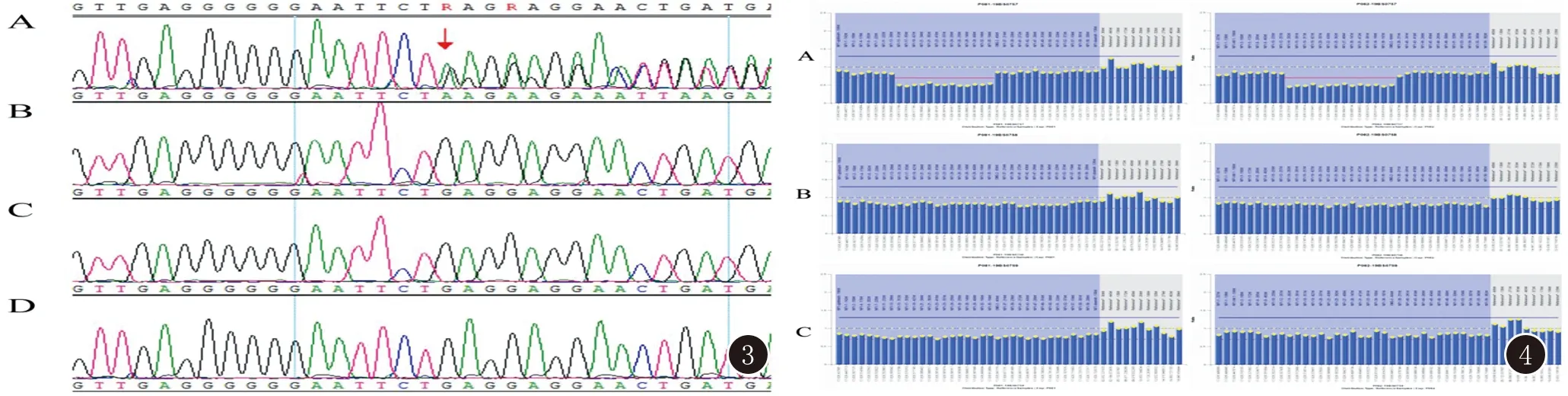
图3 患者2家系突变结果图 A为患者2患者NF1基因c.4064delC;图B、C、D分别为患者之兄、之父、之母未见相应位点变异 图4 患者3家系突变结果图 A为患者3患者NF1基因14-36号外显子杂合缺失;图B、C分别为患者之父、之母NF1基因14-36号外显子未见缺失
3 讨论
NF1的诊断主要依据1988年美国国立卫生研究院(NIH)制定的I型神经纤维瘤病的诊断标准[10]。该病典型的临床特征是:出现于腋窝、腹股沟或全身的皮肤色素沉着-牛奶咖啡斑、多个离散的良性肿瘤-皮肤神经纤维瘤和虹膜中的Lisch结节。I型神经纤维瘤病是一种由NF1基因突变引起的常染色体显性遗传病。NF1能够影响包括脑和脊髓在内的所有神经。弥散性神经纤维瘤多为良性,但其增长能够侵入周围组织从而影响其功能或者引起患者容貌缺陷。丛状神经纤维瘤易发展成恶性外周神经鞘瘤(或神经纤维肉瘤)。另外一种可能的恶性肿瘤是嗜铬细胞瘤。早期进行性退化,学习困难和认知障碍导致这些患者严重的社会障碍[11]。NF1的临床表型是进行性的,甚至在一个家族中也可能有所不同[12]。Zhang等[13]对100个有NF1临床特征的家系进行检测,发现89%具有NF1基因突变,而符合NIH诊断标准的NF1患者中,93%发现NF1基因突变。本文中3例患者均有不同程度的牛奶咖啡斑,患者1右胸有明显的肿物,患者2腋窝、腹部、腹股沟有雀斑,3例患者均检测到NF1基因的突变。患者3只有较大的咖啡斑,没有其它表型,可能是因为其年龄尚小,未完全表现出其它表型。
据统计NF1基因的自发突变率为50%[2],Nemethova等[9]对86例I型神经纤维瘤病患者家系进行研究,发现78例患者存在NF1基因的突变,其中68个患者存在NF1基因小的突变,39个为新发突变,自发突变率为50%,移码突变占42.3%,无义突变占14.1%,错义突变占12.8%,剪接突变占11.5%。Yao等[14]对95个I型神经纤维瘤病家系进行高通量测序及Sanger测序进行验证,发现77.9%为自发突变。Zhu等[15]应用DNA测序、MLPA及cDNA测序等技术对32个符合NIH诊断标准的I型神经纤维瘤病患者进行NF1基因突变检测。发现30例(93.8%)患者存在NF1基因突变,包括10个(33.3%)移码突变,4个(13.3%)错义突变,6个(20.0%)无义突变和2个(6.7%)剪接位点突变。此外还发现了8个(26.7%)涉及一个以上外显子的缺失突变。郎小乔等[16]对三例散发I型神经纤维瘤病患者进行NF1基因检测,发现三种新发突变,分别为39号外显子碱基G缺失(c.5635 del G),47号外显子大片段碱基缺失(c.7041~7062+4 del),21号外显子碱基A、C缺失(c.2714-2715 del AC)。
NF1突变谱包括错义/无义(27.7%)、剪接(16.3%)、微缺失(26.9%)、微插入(11.1%)、总缺失(>20 bp;13.3%)、总插入(>20bp;2.0%)和复杂重排(0.6%)等[8]。本文中3例I型神经纤维瘤病患者均检测到NF1基因突变,且均为自发突变,其中2例为外显子缺失突变,1例为无义突变,与现有报道的突变率相差较大,可能是因为我们收集的样本量过少所致。也可能是在甘肃地区缺失突变是常见的变异类型,是由于地域性差异造成的,这有待于继续收集病例进一步验证。
总之,我们通过对来自甘肃省妇幼保健院医学遗传中心的3例I型神经纤维瘤病患者进行高通量测序,将检测出的变异在先证者及其父母中进行MLPA以及一代测序验证,发现2个新发突变位点,丰富了NF1基因突变谱。
