H杂质在Mo-Ta晶格中成键机制的研究
2021-04-08蔡宗坚
蔡宗坚,刘 睿
H杂质在Mo-Ta晶格中成键机制的研究
蔡宗坚1,刘 睿2
(1. 广州工商学院,广东 广州 510000;2. 山东师范大学物理与电子科学学院,山东 济南 250358)
Mo-Ta合金作为一种有高熔点、抗辐照等特点的材料,被视为聚变工程中制造第一壁的潜在材料。目前有关Mo-Ta合金与H杂质相互作用的研究还不完善,本文研究了H杂质在体心立方Mo53Ta1合金中间隙位置的成键机制。通过基于密度泛函的第一性原理方法,通过Material Studio的CASTEP(Cambridge Serial Total Energy Package)模块计算了Mo53Ta1-H体系的溶解能,态密度与布居数。溶解能计算结果表明:H原子在Mo-Ta合金中更倾向于存在与四面体间隙位置;通过分波态密度与布居数,分析了H、Mo、Ta之间不同电子轨道间电子转移情况与不同原子间的成键情况。得出Mo、Ta间为金属键,H与Ta和Mo之间存在共价作用,且H与Ta之间的共价作用更强。
第一性原理;面向等离子体材料;Mo-Ta合金;氢杂质;成键机制
能源是人类社会赖以发展的基础。随着化石燃料逐渐枯竭,受控热核聚变被认为是解决能源危机的永久方案[1]。受控热核聚变装置的关键问题之一是解决面向等离子体的第一壁材料(Plasma Facing Materials,PFMs)的选择问题[2,3]。PFMs一般是第一壁材料、限制器以及偏滤器的总称,它们的共同特点就是在聚变堆工作时会直接面对高温等离子体。为应对聚变堆内严酷的工作环境,PFMs需具有低溅射、高熔点、低氢(氚)滞留等特点[4-6]。PFMs分为高Z(原子序数)和低Z材料,低 Z材料如铍或碳基材料可以减少杂质对等离子体稳定性的影响,多用来制作靶板和限制器[5];当边缘等离子体温度低,PFMs的溅射产额不足以威胁等离子体时,可使用高Z材料,如钨及其合金等高熔点金属[7]。聚变堆工作时产生的等离子流辐照会对第一壁材料造成损害,巨大的H、He通量溅射刻蚀表面原子,对PFMs的结构和性能造成重大影响[8]。目前有关H杂质与聚变堆第一壁材料相互作用的研究主要集中在最常用的第一壁材料钨及其合金上,例如钨钛合金[9]以及H与纯钨晶格中空位的相互作用[10]。低能量、高通量H/He等离子体辐照会使钨表面的微观结构产生的气泡[11,12]。
钼(Mo)和钽(Ta)是典型的高熔点过渡金属。其中钼的熔点高达2 898 K,有着高温高强度,高温下良好的抗张强度、抗蠕变强度和良好的耐热性[13]。钽的熔点更是高达3 290.15 K。钼及其合金具有熔点高、抗等离子体冲刷能力强、溅射产额低、高温强度高、热导率高等优点,并且热膨胀系数和W、C比较接近,因此被看作新型PFMs[14,15]。Cottrell[16]还指出用Mo或Ta替代钨作为第一壁材料可以有效避免因14 MeV中子辐照引起的相变导致的第一壁破裂。Mo-Ta合金是难熔体心立方(body centered cubic,bcc)合金[17],其熔点理论计算值为2 783[18],实验值为3 008 K[19]。除此之外,William Huhn等人的总结钼(Mo)、铌(Nb)、钨(W)、钽(Ta)组成的二元合金的生成焓时指出Mo-Ta合金的生成焓最低,为-186 meV/atom[20],这在其他几个难熔合金(Mo-Nb、Mo-W、Nb-Ta、Ta-W)的生成焓中是最低的,这意味着它相比其他Mo、Ta的合金具更稳定,有更较强的键合作用,是制作面向等离子的第一壁材料的不错选择。
目前面向等离子体第一壁材料与H杂质相互作用研究中,有关杂质H与Mo及其合金的研究目前还不完善。北京航空航天大学团队使用第一性原理的方法,通过对Mo晶格施加外部应力来研究应变对Mo中H杂质溶解的影响[21]。Yang等人使用Ta及其他过渡金属原子替换Mo晶格中的Mo原子并引入点缺陷,来研究过渡金属-缺陷-H原子的相互作用[22]。本文将基于第一性原理研究H杂质与Mo-Ta晶格之间的相互作用结构、溶解能、态密度、成键性质,以进一步理解H杂质在Mo-Ta合金中的行为,为面向等离子体材料选取以及设计工作提供有益的数据。
1 模型与计算方法
1.1 模型的建立
在任何温度下,Mo与Ta都能完全互溶,钼与周期系中的ⅤB、ⅥB族难熔金属(V、Nb、Ta、Cr、W)形成连续固溶体,Mo、Ta都是体心立方金属,Mo和Ta所形成的Mo-Ta合金一般是有序置换的置换固溶体,并且也为体心立方结构[23]。在Mo-Ta合金中,由于Mo和Ta可能具有多种不同的成分比例,为了更好的表现出H原子在Mo-Ta合金中的迁移特性并且尽可能的简化计算,需要选取足够大、简单且具有良好对称性的超胞作为研究对象,因此本文选取3×3×3的超胞进行研究,计算所采用的Mo53Ta1模型以3×3×3的体心立方Mo超胞为基础,以一个Ta原子替换该超胞中的一个Mo原子而形成。为了使体系满足高度对称性以简化计算,该替换位置选择为3×3×3的体心立方超胞的最中心位置,如图1所示。
图1 3×3×3 Mo53Ta1晶格
1.2 间隙位置
H原子与Mo-Ta合金的相互作用结构属于间隙相,按照间隙类型可以分为八面体型间隙相、三棱柱型间隙相、十面体型间隙相等[24]。间隙位置通常是晶体点阵中最大空隙的位置。对于Mo-Ta合金这种具有体心立方点阵结构的金属,其间隙相的间隙位置主要是八面体间隙位置(octahedral interstitial sites,OIS)和四面体间隙位置(tetrahedral interstitial sites,TIS)。八面体间隙位置是八面体型间隙相中由6个金属原子构成的八面体的中心位置;四面体间隙位置则是4个不在同一平面的金属原子构成的四面体的中心位置。相比于八面体间隙,四面体间隙是完全对称的,并且若围成间隙的金属原子完全相同,四面体间隙更大。间隙原子的引入通常会导致晶体的局部周期性遭到破坏,使晶体产生膨胀[25]。对于3×3×3的体心立方合金,其八面体间隙位置坐标为:(0.500,0.500,0.333)、(0.333,0.500,0.333)、(0.167,0.500,0.333)、(0.500,0.500,0.000)、(0.333,0.500,0.000)、(0.167,0.500,0.000)、(0.000,0.500,0.000);四面体间隙位置坐标为:(0.417,0.500,0.333)、(0.250,0.500,0.333)、(0.166,0.417,0.333)、(0.083,0.500,0.333)、(0.417,0.500,0.000)、(0.250,0.500,0.000)、(0.166,0.417,0.000)、(0.083,0.500,0.000)。

图2 (a)3×3×3 Mo53Ta1-H(TIS);(b)3×3×3 Mo53Ta1-H(OIS)晶格。其中灰色为Mo原子,白色为Ta原子,黑色为H原子
1.3 计算方法
本文研究采用Materials Studio 2017(17.1.0.48)的CASTEP(Cambridge Serial Total Energy Package)模块计算[26]。交换关联泛函采用广义梯度近似(Generalized Gradient Approximation,GGA)的PW91泛函,使用超软赝势(Ultra-Soft Pseudo-Potentials,USPP)[27],截断能为300 eV,k点3×3×3网格[28],能量收敛精度为1×10-5eV/ atom,最大力收敛精度为0.3 eV/nm,最大应变收敛精度为0.05 GPa,最大位移收敛精度为0.01 nm。
此外,我们使用Mulliken和Hirshfeld方法计算了电荷布居数。Mulliken布居是最早的原子电荷计算方法,基于波函数,它将轨道中的电子分为居于在原子轨道中的电子和轨道重叠区的电子两部分,并将重叠区的电子平均分给相关原子轨道。Hirshfeld布居是基于电荷密度来定义原子电荷,并按权重将重叠区电荷进行分配[29]。
2 结果与讨论
2.1 结构优化
H原子介入Mo-Ta合金,尤其是Mo53Ta1的不同位置的稳定性差异与H原子与Ta原子的相互作用有很大关系。在Mo53Ta1中Ta原子的浓度较低,必须使H原子与Ta原子充分作用才能体现出Mo-Ta合金与H原子的作用效果。因此为了更好体现H原子介入Mo-Ta合金四面体间隙位置和八面体间隙位置的差异,选择超胞中距离Ta原子最近的八面体间隙和四面体间隙作为比较H原子在Mo53Ta1中最稳定位置的模型[30-31],其坐标分别为(0.500,0.500,0.333)和(0.417,0.500,0.333)。
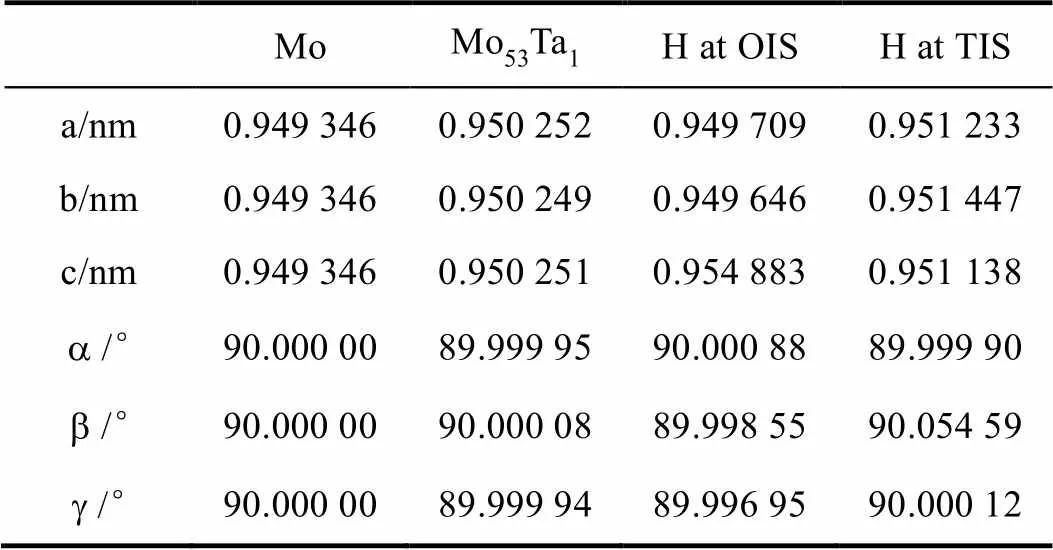
表1 结构优化后的晶格常数
原子替换与杂质介入均会使晶格发生畸变,这种变化主要体现在晶格的边长上,晶面夹角的变化很小,故探讨体积变化时可以将a、b、g视作保持90°不变。通过计算各体系的的体积变化率发现,H原子在OIS位置时体系的体积变化率最大,为0.65%,其次是TIS位置时的0.61%。这一体积变化率与Fukai[32]提到的TIS间隙体积比OIS大相吻合,TIS更大的体积为H原子提供了更合适的空间,使得体积变化率更小。一般来说,晶格发生畸变需要吸收能量,从而导致整个体系能量上升,稳定性下降。相比与结构优化后的Mo53Ta1晶格,H原子介入OIS位置的结构在a轴和b轴方向有着微小的“压缩”效果,而在[0 0 1]方向有着明显的“拉伸”效果。虽然计算的所有体系的晶格畸变都不是各向同性的,但是H原子介入OIS位置的结构的不同方向畸变的差异显然要大很多。H原子的坐标选为(0.500,0.500,0.333)的缘故,该位置在连线沿[0 0 1]方向的Ta原子与Mo原子的中间,并且与这两个原子的距离比围成八面体的其他四个原子的距离近。在这里插入H原子后,弹性作用使得这两个原子的间距增大到了35.33 nm,而未插入前这个间距只有31.76 nm。因此在这个位置插入H原子后会导致沿[0 0 1]方向的晶格常数变大的程度明显大于其他两个方向上晶格常数变化的程度。
2.2 溶解能
溶解能是原子溶入合金时整个体系所吸收或释放的能量,它能反映体系的稳定程度,从而进一步得出H原子在Mo-Ta合金中占据位置的倾向。我们通过计算H原子在Mo-Ta合金中不同间隙位置的溶解能来确定H原子所处的间隙位置。通过公式(1)进行计算。

其中为溶解能,为含H体系的总能,为不含H体系的总能,为单个H原子的能量。其中计算得到Mo53Ta1的能量为-102 840.68 eV,H处于OIS的能量为-102 855.61 eV,H在TIS能量为-102 856.09 eV,二分之一H2能量为-15.828 eV。最后由式(1)计算出H原子在OIS和TIS两个位置时体系的溶解能分别为0.898 eV和0.418 eV,这说明H原子在Mo-Ta合金中倾向于占据TIS位置。

图4 Mo53Ta1-H体系中原子的分态密度图,(1)为H处于OIS位置,(2)为H处于TIS位置。其中-s,-p,-d分别为电子轨道,-n,-f分别为离中心Ta原子最近和最远。
2.3 态密度分析
分波态密度显示H原子的态密度具有很强的局域性,H原子与周围其他原子的杂化主要发生在能量约-8 eV处。H原子的1s轨道与Ta原子的5p轨道在该能量范围内发生了明显的sp杂化,但H原子在TIS位置相比于在OIS位置,此处H原子的1s轨道与Ta原子的5p轨道的杂化显然要弱一些。除此之外,在该能量区间内,Ta原子的6s和5d轨道也存在部分贡献。与H原子最邻近的Mo原子与H原子也在这个能量范围内发生了sd杂化,这使得相比于离H原子最远的Mo原子其d轨道态密度在大约2 eV处有略微的下降,并且对于TIS位置的H原子来说,并没有与最邻近Mo原子之间表现出相比OIS位置更强的杂化作用。此外与H原子最远的Mo原子没有与H原子发生杂化,说明H原子的与该晶格中的其他原子的成键作用有一定的局域性。Ta原子的态密度在费米能级两侧存在着态密度峰并且费米能级处态密度不为零,即在能级中存在赝能隙,这可以说明H原子与Ta原子形成了较强的共价键。
溶解能的计算结果指出在H原子在Mo-Ta合金中倾向于占据TIS位置,该位置更加稳定,意味着H原子与周围原子的杂化理论上应该更强。出现这种情况的原因可能是与H原子最邻近的其他原子的数目不同。在OIS位置的优化体系中,H原子与Ta原子以及最邻近的Mo原子的距离分别是0.181 nm和0.172 nm,这两个距离均比TIS位置优化体系中H原子与Ta原子以及最邻近Mo原子的距离小(0.195 nm和0.183 nm),但是TIS位置处H原子的最邻近Mo原子却有三个。因此虽然在TIS位置时H原子与其他单个原子的杂化不强,但与周围原子的电子云重叠区域更大,这可能导致了H原子在该位置时更加稳定。
2.4 布局分析
由电负性大小H>Mo>Ta可知,H原子周围会汇聚大量负电荷,H周围的Mo会失电子,但Ta原子周围也集中了大量的负电荷,这从电负性的角度有一些差异,这可能是因为H-Ta原子间的键重叠布居数较大,存在很强的共价作用,使H周围的电荷与Ta共用导致。H获得的电子大部分来自于Mo原子的4p轨道。由表2,表3可知,Ta原子的5p、5d、6s轨道全部得到了额外电子,并且主要体现在5d轨道上。两个体系中与H原子邻近的Mo原子基本上都带有较多的正电荷,表明H原子与这些Mo原子之间存在着比较强烈的吸引作用。TIS位置H原子周围带正电的Mo原子更多,说明其与周围Mo原子有更强的键合作用,因此该体系更加稳定,这与前文给出的结论相符。
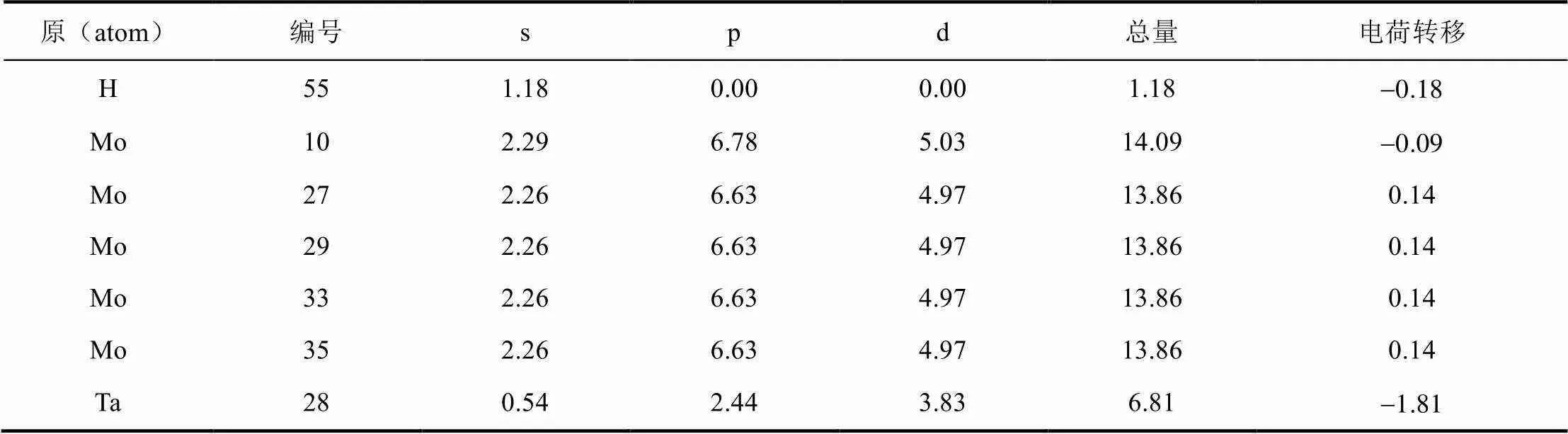
表2 H在OIS位置马利肯布居数

表3 H在TIS位置马利肯布居数
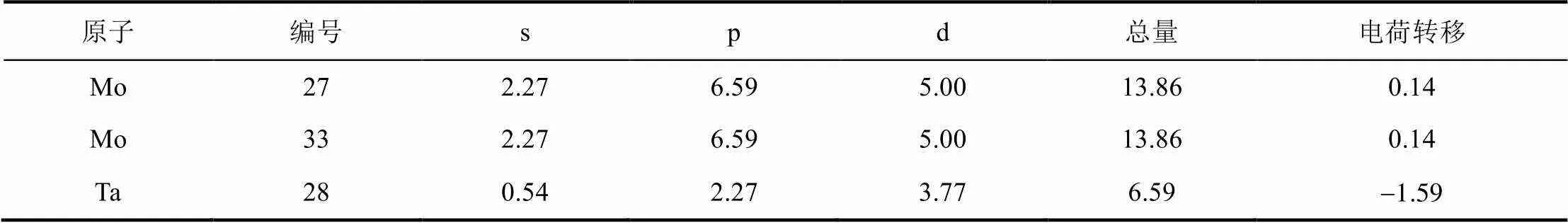
续表

表4 H在OIS位置原子间键重叠布居数

表5 H在TIS位置原子间键重叠布居数

表6 H在OIS位置赫氏布居数

表7 H在TIS位置赫氏布居数
其中H原子位于OIS位置的体系中H原子与Ta原子之间的共价键更强。在H原子介入OIS位置的体系中,H原子与Mo10之间也存在着一定共价作用,但是键的强度不如H原子与Ta原子之间共价键的强度。H原子之所以能在此处形成这两个共价性比较强的键,是因为这个八面体间隙位置前后左右是高度对称的,H原子只能在Ta和Mo10之间的狭小空隙内驰豫,这导致H原子与这两个原子之间的电子云重叠密度比较大,键具有较高的共价性。H原子与Mo27、Mo28、Mo32、Mo34之间存在程度较高的反键态结合,在H原子介入TIS位置的体系中,H原子与周围多数Mo原子也是反键态结合,但其强度不如OIS位置的体系的反键,电子填入反键轨道会使体系的稳定性降低,因此H原子在TIS位置时体系更稳定。表6,表7所示的Hirshfeld布居与Mulliken布居结果相似,不再赘述。
3 结论
本文通过基于密度泛函理论的第一性原理方法计算了Mo53Ta1合金的结构以及杂质H原子在该结构中的部分行为。通过对八面体间隙位置(OIS)和四面体间隙位置(TIS)体系进行了结构优化以及能量计算,推断出H原子在Mo53Ta1合金中倾向于占据的间隙位置,并进一步通过态密度、布居数研究了H原子在间隙位置与Mo53Ta1合金的成键性质,主要结论如下:
(1) Ta原子替换和H杂质原子介入都会对晶体的晶格造成不同程度的畸变。杂质原子介入会导致Mo-Ta合金的晶格常数增大。H原子介入Mo-Ta合金OIS位置后其对最近邻两个原子的弹性作用会使该体系的晶格沿[0 0 1]方向发生明显的伸展。H原子介入OIS位置后晶格体积的膨胀率也更大,意味这该结构的稳定性可能较差;
(2)通过计算H在OIS和TIS位置体系的溶解能得H原子在Mo-Ta合金中倾向于占据TIS位置;
(3)通过态密度与布居分析显示,相比于Mo原子,H原子与Ta原子之间存在更强的共价作用。
[1] Jian-Cheng Liu.The Controlled Thermonuclear Fusion Hydrogen Energy-A Permanent Solution to the World's Energy Supply.2010,21(2):93-96.
[2] 丁厚昌,黄锦华.受控核聚变研究的进展和展望[J].自然杂志,2006(03):143-149.
[3] 黄学龙,信敬平,毛小东,等.1.61dpa/300 ℃中子辐照后CLAM 钢的硬化和脆化行为[J].核科学与工程,2018,38(2):225-230.
[4] 许增裕.聚变材料研究的现状和展望[J].原子能科学技术,2003(S1):105-110.
[5] Ulrich Samm.Controlled thermonuclear fusion at the beginning of a new era[J].Contemporary Physics,2003,44(3).
[6] 丁孝禹,李浩,罗来马,等.国际热核试验堆第一壁材料的研究进展[J].机械工程材料,2013,37(11):6-11.
[7] 周张健,钟志宏,沈卫平,等.聚变堆中面向等离子体材料的研究进展[J].材料导报,2005(12):5-8+12.
[8] 吕广宏,罗广南,李建刚.磁约束核聚变托卡马克等离子体与壁相互作用研究进展[J].中国材料进展,2010,29(07):42-48.
[9] D.Y.Jiang,C.Y.Ouyang,S.Q.Liu.The effect of titanium(Ti)doping on hydrogen incorporation in tungsten(W):First-principles calculations[J].Fusion Engineering and Design,2017,121.
[10] Xiao-Chun Li,F.Gao,Guang-Hong Lu.Molecular dynamics simulation of interaction of H with vacancy in W[J].Nuclear Inst.and Methods in Physics Research,B,2009,267(18).
[11] Ye M Y,Takamura S,Ohno N.Study of hot tungsten emissive plate in high heat flux plasma on NAGDIS-I[J]. Journal of Nuclear Materials,1997,241(1):1243-1247.
[12] Ye Minyou. Effects of Low Energy and High Flux Helium/ Hydrogen Plasma Irradiation on Tungsten as Plasma Facing Material[J].Plasma Science &;Technology,2005(03):2828-2834.
[13] 中国有色金属工业协会.中国钼业[M].北京:冶金工业出版社,2013:4-6.
[14] 张小锋,刘维良,郭双全,等.聚变堆中面向等离子体材料的研究进展[J].科技创新导报,2010(03):118-119.
[15] 淡新国,黄先明,郭让民,等.聚变堆中面向等离子体的钼板材制备工艺研究[J].中国钼业,2014,38(01):50-53.
[16] G.A.Cottrell.Sigma phase formation in irradiated tungsten,tantalum and molybdenum in a fusion power plant[J]. Journal of Nuclear Materials,2004,334(2).
[17] Blum V,Zunger A.Prediction of ordered structures in the bcc binary systems of Mo,Nb,Ta,and W from first- principles search of approximately 3,000,000 possible configurations[J].Physical Review B Condensed Matter & Materials Physics,2005,72(2):104.
[18] De Coss,R.,Aguayo,A.,& Murrieta,G.(2000). First- Principles Calculations of Electronic Structure and Structural Properties for MoV,MoNb,and MoTa.MRS Proceedings,646,N5.33.1.
[19] Huhn W,Widom M.Prediction of A2 to B2 Phase Transition in the High Entropy Alloy Mo-Nb-Ta-W[C]//APS March Meeting 2014.American Physical Society,2014.
[20] Moseley P T,Seabrook C J.The crystal structure of β-tantalum[J].Acta Crystallographica Section B Structural Crystallography and Crystal Chemistry,1973,29(5):1170-1171.
[21] Han,Quan-Fu,et al. “Applying isotropic strain on Mo to predict H solution behaviors for nuclear energy application: Temperature dependence.” International Journal of Hydrogen Energy 43.7(2018):3750-3760.
[22] Yang,Kun Jie,et al. “First-principles simulation of H interacting with transition elements in molybdenum for nuclear material application.” Journal of Nuclear Materials 541(2020):152437.
[23] 李玉清,刘锦岩.高温合金晶界间隙相[M].北京:冶金工业出版社,1990:51-85.
[24] 陈继勤,陈敏熊,赵敬世.晶体缺陷[M].浙江:浙江大学出版社,1992:3-7.
[25] Payne M C,Arias T A,Joannopoulos J D.Iterative minimization techniques for ab initio total-energy calculations:molecular dynamics and conjugate gradients[J].Reviews of Modern Physics,1992,64(4):1045-1097.
[26] Segall M D,Lindan P J D,Probert M J,et al. First- principles simulation:ideas,illustrations and the CASTEP code[J].Journal of Physics Condensed Matter,2002,14(11):2717-2744.
[27] Vanderbilt.Soft self-consistent pseudopotentials in a generalized eigenvalue formalism.[J].Physical review.B,Condensed matter,1990,41(11).
[28] Monkhorst H J,Pack J D.Special points for Brillouin-zone integrations[J].Physical Review B,1976,13(12):5188-5192.
[29] 卢天,陈飞武.原子电荷计算方法的对比[J].物理化学学报,2012,28(01):1-18.
[30] Y.Fukai,The Metal-Hydrogen System,second ed.,springer,Berlin,2005,p.16.
[31] Qin,Jiayao,et al.“First-principle investigation of hydrogen solubility and diffusivity in transition metal-doped vanadium membranes and their mechanical properties.”Journal of Alloys and Compounds 805(2019):747-756.
[32] Y.Fukai. The Metal-Hydrogen System:Basic Bulk Properties. Berlin,Heidelberg,Springer.2005.
Study on the Bonding Mechanism of H Impurity in the Mo-TA Lattice
CAI Zongjian1,LIU Rui2
(1. Guangzhou College of Technology and Business, Guangzhou of Guangdong Prov. 510000, China;2. School of Physics and Electronics, Shandong Normal University, Jinan of Shandong Prov. 250358, China)
As a kind of material with high melting point and anti-irradiation, the Mo-TA alloy is regarded as a potential material for making the first wall in fusion engineering. At present, the study on the interaction between the Mo-Ta alloy and H impurity is not perfect. In this paper, the bonding mechanism of H impurity in body centered cubic Mo53Ta1 alloy is studied. The solution energy, density of states and population of Mo53Ta1-H system were calculated by CASTEP module of the Material Studio. The calculated results of solution energy show that H atoms are more likely to exist in the tetrahedron interstitial sites in the Mo-Ta alloy. The electron transfer between different electron orbitals of H, Mo and Ta and the bonding between different atoms are analyzed by means of density of states and population. It is concluded that Mo and Ta are metal bonds, H has covalent interaction with Ta and Mo, and the covalent interaction between H and Ta is stronger
First principle; Plasma facing materials; Mo-Ta alloy; H impurity; Bonding mechanism
TL99
A
0258-0918(2021)06-1332-09
2021-03-05
广州工商学院人文社科类校级科研项目《习近平强军思想引领下高校国防教育中大学生革命战斗精神的培育与研究》(KA202015)
蔡宗坚(1983—),广东阳江人,讲师,学士,现主要从事军事学和核科学方面研究
