Gd2SiO5纳米粉体的并流化学共沉淀法合成
2021-04-08王雅雷武囡囡刘怀菲刘如铁程慧聪
刘 蓉, 王雅雷*, 武囡囡, 刘怀菲, 刘如铁, 程慧聪
(1.中南大学 粉末冶金研究院,长沙 410083;2.中南林业科技大学 材料科学与工程学院,长沙 410004)
碳化硅纤维增强碳化硅复合材料(SiCf/SiC)具有低密度、高比强、高比模、耐高温等优点,作为航空发动机热端部件不仅可减少冷却需求、改进热结构部件的耐久性,还可减轻发动机质量、提高航空发动机推力和效率,已成为高推重比航空发动机高温热材料的发展方向[1-2]。然而,在航空发动机应用环境下,SiCf/SiC复合材料存在易受水氧腐蚀及CMAS(CaO-MgO-Al2O3-SiO2)腐蚀问题,致使材料表面稳定性及性能急剧恶化[3-5]。目前最有效的途径是在SiCf/SiC复合材料构件表面制备环境障涂层(environmental barrier coatings,EBC)来提高材料及部件的环境稳定性[6]。
EBC涂层主要用来抵御航空发动机燃气环境对SiCf/SiC复合材料构件的腐蚀,同时兼具阻断和愈合裂纹、孔隙的作用,是确保SiCf/SiC复合材料构件长时稳定服役的关键[7]。稀土硅酸盐具有高熔点( > 1800 ℃)、极低的高温氧渗透率、低热导率、低热膨胀系数、低的硅活度、低模量、低饱和蒸气压以及优良的高温化学稳定性、抗水氧腐蚀性能和抗CMAS腐蚀性能,且在高速燃气环境中具有较低的挥发率,是新一代EBC涂层体系的首选材料,也是未来高性能EBC涂层发展的重要方向[8-9]。稀土硅酸盐包括稀土单硅酸盐(RE2SiO5)和稀土焦硅酸盐(RE2Si2O7)两类。在稀土单硅酸盐中,RE原子半径较大时(RE = La、Ce、Pr、Nd、Pm、Sm、Eu、Gd)易形成低温相结构(X1-RE2SiO5);RE原子半径较小时(RE = Dy、Ho、Er、Sc、Tm、Yb、Lu),稀土单硅酸盐则易形成高温相结构(X2-RE2SiO5)[10-11]。稀土单硅酸盐中,Gd2SiO5熔点高达1950 ℃,且因具有独特的半满带核外电子分布,仅以X1相单硅酸盐结构存在,拥有良好的高温结构稳定性[12]。另外,在1400 ℃水氧耦合环境下,Gd2SiO5表现出优异的抗腐蚀性能[11]。李其连等[13]利用真空等离子喷涂制备了以Gd2O3掺杂Si为底层、Gd2SiO5为中间层、Yb2SiO5为面层的EBC涂层,该涂层表现出良好的相稳定性和抗热震性能,且层间结合良好。因此,Gd2SiO5作为EBC涂层材料具有良好的应用前景。
对于EBC涂层的制备和综合性能而言,稀土硅酸盐材料的纯度、粒度、形貌等特征极其关键[14],且主要取决于粉体材料的制备方法。目前,Gd2SiO5粉体大多采用固相反应法合成[15-16]。Nasiri等[17]采用固相法制备了Gd2SiO5粉末,该材料在927 ℃的热导率仅为1.6 W•m–1•K–1,且随温度升高并未明显增加,表现出良好的稳定性。王楠等[18]以Gd2O3、SiO2为原材料,采用固相法制备了Gd2SiO5粉体。其中,1350 ℃煅烧合成的Gd2SiO5粉体中明显含有Gd2Si2O7、Gd2O3和SiO2等杂质相,且同时伴有微量非晶相存在。后来通过提高烧结温度至1500 ℃才获得相对较纯的Gd2SiO5粉体。Gd2O3和SiO2均为高熔点物质,在1500 ℃的高温条件下仍会以固态形式存在,通常会采用添加助溶剂的方式来促进反应[19]。固相法制备硅酸盐粉体虽然工艺成熟,但制备过程易引入杂质,且存在所得粉体颗粒较大、粒径控制困难、粉体内部元素分布均匀性较差等问题[20]。化学共沉淀法制备粉体材料可实现原料分子、原子级的混合,易于获得成分均匀的超细粉体,且具有设备简易、工艺简单可控、粉体材料纯度高、合成温度低等优势。中南大学在原有传统化学共沉淀方法的基础提出了一种新型的并流共沉淀法[21-22],并成功合成了DySZ和Yb2SiO5纳米粉体材料。本研究便是采用并流共沉淀法制备Gd2SiO5粉体材料,系统研究Gd/Si摩尔比、煅烧温度、反应体系pH等参数对Gd2SiO5粉体材料物相组成与微观形貌的影响,初步探讨Gd2SiO5粉体材料的合成机理。
1 实验材料及方法
1.1 原材料
氧化钆(Gd2O3,99.99%(质量分数, 下同)),湖南稀土金属材料研究院;盐酸(HCl,36%~38%),国药集团化学试剂有限公司;正硅酸乙酯(C8H20O4Si,TEOS,SiO2含量 ≥ 28.0%),西陇化工股份有限公司;无水乙醇(C2H5OH,≥ 99.7%),天津市恒兴化学试剂制造有限公司;氨水(NH3•H2O,25%~28%),湖南汇虹试剂有限公司。
1.2 材料制备
首先,称取一定量Gd2O3粉末,加入沸腾的稀释盐酸溶液中形成GdCl3溶液,冷却后通过添加去离子水调节溶液中的Gd3+浓度至1 mol/L,该溶液标记为阳离子溶液A;然后,根据设计的Gd/Si摩尔比(20∶9~20∶12)量取一定体积的TEOS,按照TEOS∶无水乙醇∶去离子水体积比为1∶2∶4的比例进行混合、搅拌,得到TEOS溶液,该溶液标记为阳离子溶液B。最后,将阳离子溶液A和阳离子溶液B进行充分混合,得到阳离子溶液C。此外,将氨水和去离子水按照体积比1∶1进行混合得到沉淀剂溶液D。
在并流共沉淀过程中,首先通过在去离子水中添加氨水来调节母液pH值至设定值(8~11),然后将阳离子溶液C和沉淀剂溶液D持续加入至持续搅拌的反应母液中。其中,阳离子溶液C的流动速率保持不变,通过调整沉淀剂溶液D的流速将反应环境pH值稳定控制在目标值。沉淀过程完成后,继续保持搅拌30 min得到前驱体溶液。将得到的沉淀溶液在室温环境下陈化24 h、去离子水洗涤去除溶液中的Cl–直至pH值为7、无水乙醇洗涤、抽滤后得到前驱体湿凝胶,湿凝胶通过110 ℃干燥12 h得到Gd2SiO5前驱体。以酒精为球磨介质,利用行星式球磨机以200 r/min的速度湿磨2 h,85 ℃干燥8 h后得到Gd2SiO5前驱体粉末。最终,将前驱体粉末放入高温炉中,在1000~1300 ℃空气气氛中进行煅烧,得到Gd2SiO5粉体材料。Gd2SiO5粉体制备的主要工艺参数如表1所示。
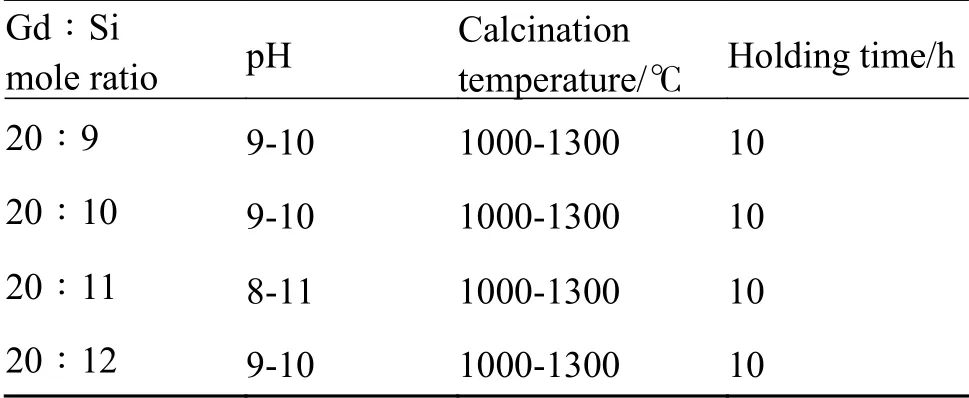
表1 Gd2SiO5粉体制备的主要工艺参数Table 1 Preparation parameters of Gd2SiO5 powder
1.3 测试与表征
采用STA449C型同步热分析仪对Gd2SiO5前驱体粉末进行TG-DSC分析,测试范围为室温~1200 ℃,升温速率为10 ℃/min,空气气氛。
采用Advance-D8型X射线衍射仪(35 kV,35 mA,0.15406 nm Cu靶Kα射线)分析Gd2SiO5粉体的物相组成,扫描速率为8 °/min,扫描范围为10~80 °。
采用Tecnai G2 F20型透射电子显微镜分析Gd2SiO5粉体的微观形貌和晶体结构,利用其配备的能量色散光谱仪(EDS)分析Gd2SiO5粉体颗粒内部的元素分布。
2 结果与分析
2.1 Gd2SiO5前驱体的热响应特征
图1所示为不同Gd∶Si摩尔比合成Gd2SiO5前驱体的TG-DSC曲线。由图1可以看到,不同Gd∶Si摩尔比合成Gd2SiO5前驱体的TG-DSC曲线变化趋势无明显差异,说明四种前驱体在转化过程中经历了类似的吸放热和转换过程。在室温~200 ℃区间,前驱体存在8%左右的失重,这主要是由前驱体粉末中物理吸附的水分子与乙醇分子受热挥发所致[23],在DSC曲线中对应明显的吸热峰。在200~500 ℃区间,前驱体的失重约为7%,DSC曲线在此区间则表现为一个明显宽泛的放热峰。这是因为,前驱体中存在一定量的烷氧基团和碳-氢基团,两种基团之间在此温度范围内发生反应生成了挥发性气体,并同时释放了一定的能量[24]。在500~900 ℃区间,前驱体自身质量没有明显变化,同时未发现明显的吸放热行为。在此阶段,前驱体内部仅发生部分碳氢基团的燃烧[25],故整体减重不明显。在900~1100 ℃区间范围内,前驱体的DSC曲线中均可见一个明显的放热峰,且同时伴随质量的下降;而当温度大于1100 ℃时,前驱体的质量保持稳定,DSC曲线中也未发现明显的吸放热现象。这说明,Gd2SiO5前驱体已经基本完成了向Gd2SiO5晶体的转化。值得注意的是,不同Gd∶Si摩尔比合成Gd2SiO5前驱体的晶化温度存在明显差异。由图1可以看出,当Gd:Si摩尔比为20∶12、20∶11、20∶10和20∶9时,前 驱 体 向Gd2SiO5转化的结晶温度分别为949.8 ℃、961.1 ℃、1025 ℃和1020.1 ℃。当Gd∶Si摩尔比值低于20∶10时,Gd2SiO5粉体的结晶温度随Si配比量的降低呈现增加的趋势;而当Gd∶Si摩尔比高于20∶10时,Gd2SiO5粉体的结晶温度则稍有下降。总体而言,采用并流沉淀法制备Gd2SiO5粉体的煅烧合成在1000 ℃左右即可完成,明显低于固相法[18]。
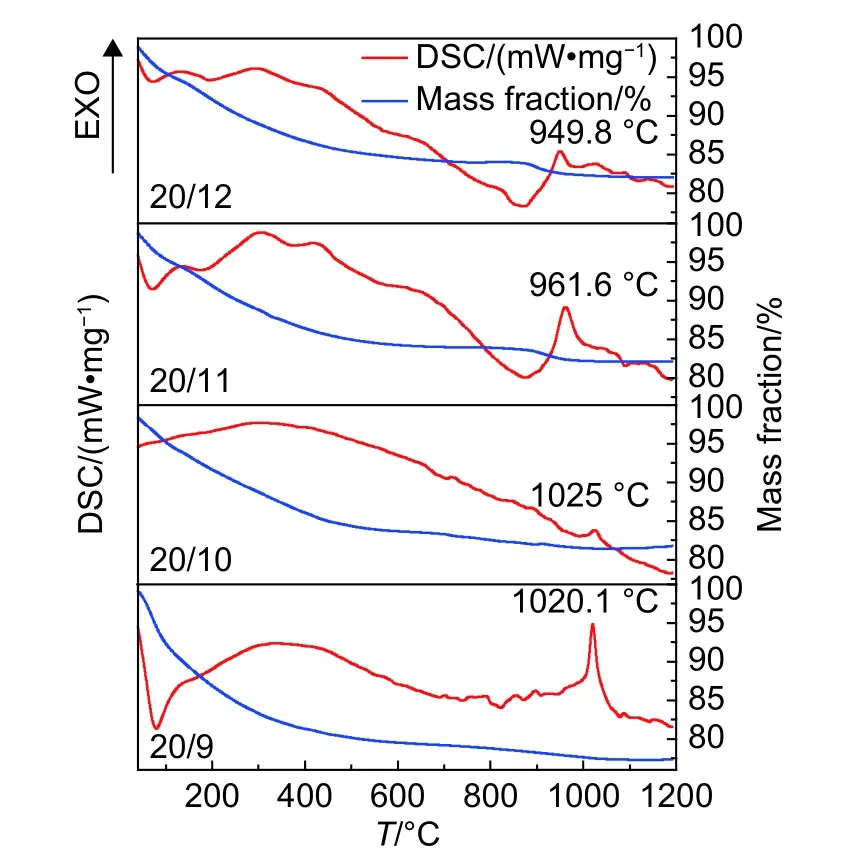
图1 不同Gd∶Si摩尔比合成Gd2SiO5前驱体的TG-DSC曲线Fig. 1 TG-DSC curves of Gd2SiO5 precursors synthesized with different Gd∶Si molar ratios
2.2 Gd2SiO5粉体的物相组成
根据Gd2O3-SiO2相图可知[26],当Gd∶Si计量比在20∶10~20∶12之间时,室温条件下所合成化合物是由Gd2O3-SiO2和Gd2O3-2SiO2组成的;而当Gd∶Si计量比高于20∶10时,室温条件下所合成化合物则是由Gd2O3和Gd2O3-SiO2组成的。图2所示为不同Gd∶Si摩尔比前驱体1100 ℃煅烧产物的XRD图谱。从图2可看出,不同Gd∶Si摩尔比合成前驱体的煅烧产物中均出现了明显的Gd2SiO5的衍射特征峰,对应标准JCPDS图卡No.74-1795。这说明在1100 ℃煅烧条件下,Gd2SiO5前驱体可以完成向Gd2SiO5晶体的转化,且与固相法相比,并流共沉淀法明显降低了Gd2SiO5的合成温度。
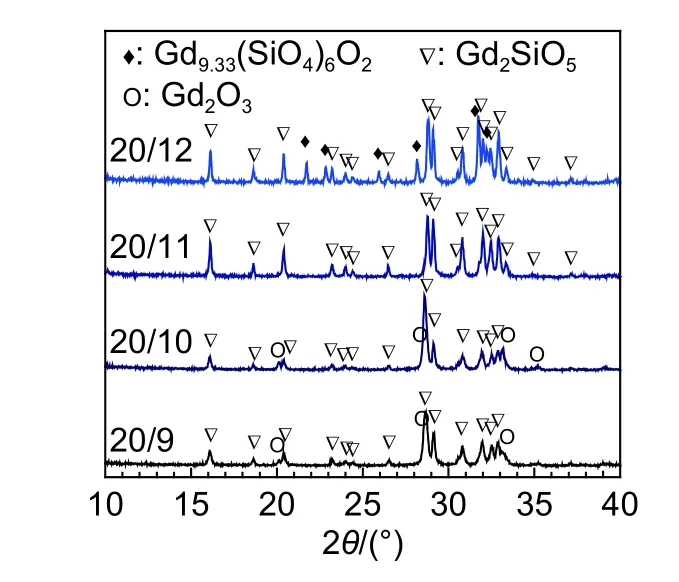
图2 不同Gd:Si摩尔比前驱体1100 ℃煅烧10 h合成产物的XRD图谱Fig. 2 XRD patterns of Gd2SiO5 precursors with different Gd:Si ratios calcined at 1100 ℃ for 10 h
另外,由图2可以明显看到,不同Gd∶Si摩尔比条件制备的Gd2SiO5粉体的物相组成存在明显差异。当Gd∶Si摩尔比为20∶12时,Gd2SiO5粉体由Gd2SiO5和Gd9.33(SiO4)6O2(PDF#38-0283)组成。当Gd∶Si摩尔比为20∶11时,所合成的Gd2SiO5粉体的XRD图谱中仅可见Gd2SiO5的特征峰,合成的Gd2SiO5粉体纯度较高。当Gd∶Si摩尔比为20∶10和20∶9时,Gd2SiO5粉体则由Gd2SiO5和少量的Gd2O3(PDF#76-0155)组成,其中当Gd∶Si摩尔比为Gd2SiO5中Gd/Si化学计量比时,所制备的Gd2SiO5粉体中生成了少量的Gd2O3相。这是因为,一方面Gd2SiO5粉体合成原料中TEOS中的SiO2含量难以精确测量,另一方面与粉体材料合成过程中Si的损失有关。值得注意的是,当Gd∶Si摩尔比为20∶12时,所合成的Gd2SiO5粉体中生成了稳定的Gd9.33(SiO4)6O2相。相关研究表明[27]:Gd9.33(SiO4)6O2相是一种具有磷灰石结构,处于Gd2SiO5与Gd2Si2O7之间的中间态物质。尽管与单斜相Gd2SiO5所属空间群不同,但Gd9.33(SiO4)6O2与Gd2SiO5晶体结构中均是由[SiO4]四面体与[GdOx]多面体组成,其中Gd在晶体结构中都具有7氧配位和9氧配位两种配位结构。吴瑞芬等[28]将Gd2O3与SiO2按照摩尔比为10∶12、9∶12和8.67∶12进行混合,采用固相法煅烧制备了Gd10-x(SiO4)6O3-1.5x(x= 0,1,1.33)陶瓷材料。另外,Yokota等[27]以Gd(NO3)3和TEOS为原材料,采用化学共沉淀法在1300 ℃合成了Ce改性Gd2SiO5材料,在Gd∶Si摩尔比偏低的条件下含有少量的Gd9.33(SiO4)6O2;并且在同样的烧结温度下通过调控Gd:Si摩尔比成功制备了较纯的Gd9.33(SiO4)6O2材料。此外,由图2还可以看到,随着合成前驱体中Gd∶Si摩尔比的降低,所合成粉体材料中Gd2SiO5衍射特征峰的峰强逐渐增加,半高宽减小,这说明前驱体中的富Si环境有助于促进Gd2SiO5晶体的形成,这与Gd2SiO5前驱体的TG-DSC分析结果是一致的。
图3所示为不同Gd∶Si摩尔比前驱体1100~1300 ℃煅烧产物的XRD图谱。由图3可知,在不同Gd∶Si摩尔比条件下,前驱体在1000~1300 ℃煅烧范围内合成Gd2SiO5粉体的物相组成没有差异。说明煅烧温度对并流共沉淀法制备Gd2SiO5粉体的物相组成没有明显的影响。另外,由图3可以发现,在所得Gd2SiO5粉体材料中,Gd2SiO5、Gd2O3、Gd9.33(SiO4)6O2三种物相在1000 ℃时就已开始结晶形成,且随着煅烧温度的提高,并未发现上述物相衍射峰的消失和相对强度的变化,这说明上述物相一旦生成便会以一种稳定的结构形式存在。翟志学[29]研究表明:Gd9.33(SiO4)6O2和Gd2SiO5都具有极高的高温相稳定性,材料合成过程中均无法通过改变烧结条件来去除主相中的第二相。此外由图3可以看到,随着煅烧温度的升高,Gd2SiO5衍射峰的强度增加、峰形更加尖锐,Gd2SiO5粉体的结晶性可以得到明显提升。
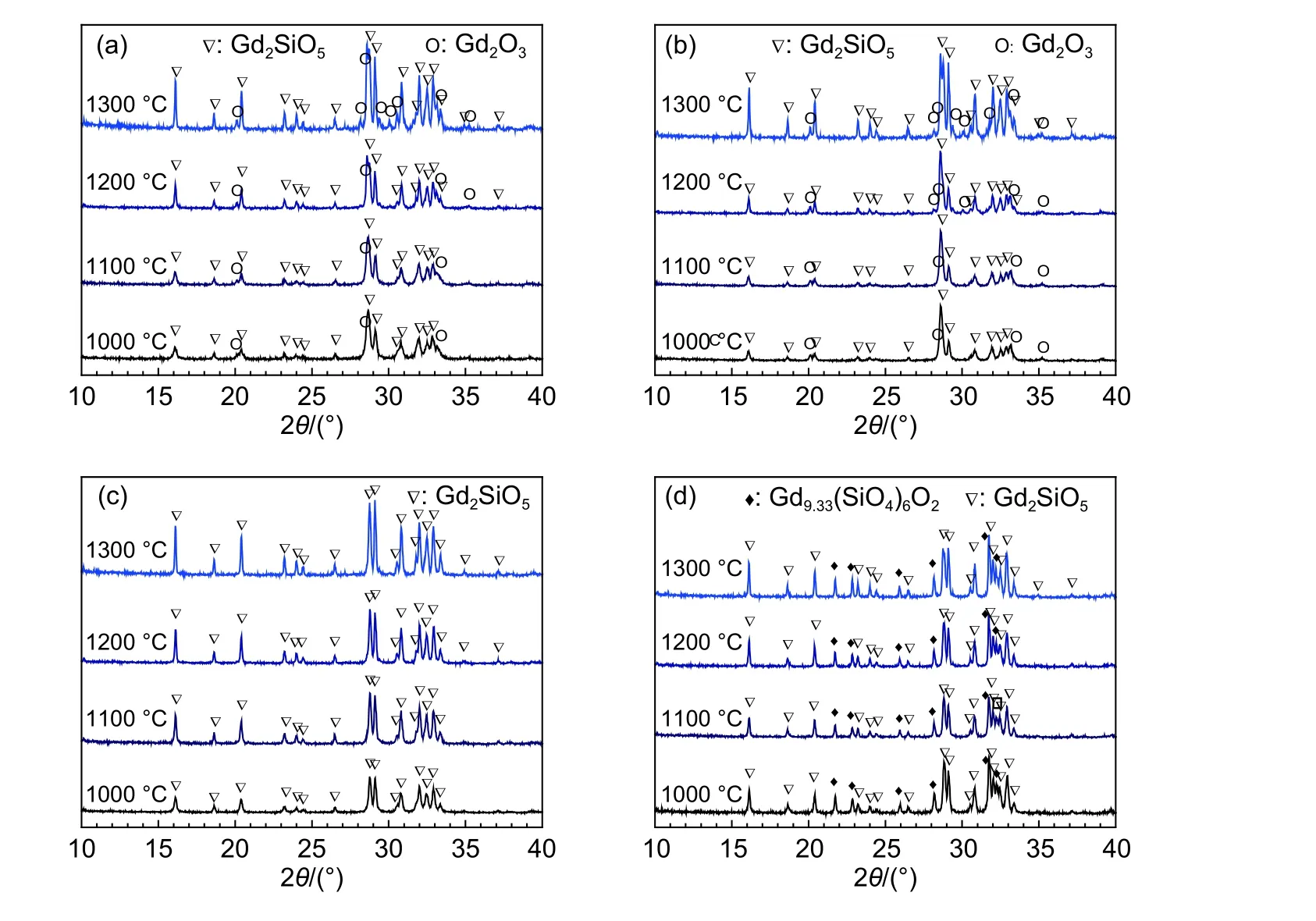
图3 不同Gd∶Si摩尔比前驱体1100~1300 ℃煅烧产物的XRD图谱 (a)Gd∶Si=20∶9;(b)Gd∶Si=20∶10;(c)Gd∶Si=20∶11;(d)Gd∶Si=20∶12Fig. 3 XRD patterns of Gd2SiO5 precursors with different Gd∶Si ratios calcined at 1100~1300 ℃ for 10 h (a)Gd∶Si=20∶9;(b)Gd∶Si=20∶10;(c)Gd∶Si=20∶11;(d)Gd∶Si=20∶12
本研究采用并流共沉淀法在1000 ℃合成具有成分稳定特征的Gd2SiO5粉体,这说明与固相法合成Gd2SiO5粉体的机理是完全不同的。在并流共沉淀法合成Gd2SiO5粉体的过程中,Gd2SiO5前驱体在煅烧过程中是从具有无定形态特征的复合盐沉淀直接转化形成Gd2SiO5和Gd9.33(SiO4)6O2,并未通过Gd2O3和SiO2作为中间产物。
在采用并流化学共沉淀法制备Gd2SiO5粉体的过程中,反应体系的pH值是一个重要的影响因素[30]。本研究通过溶度积计算得到了Gd3+沉淀所需要的pH值,溶度积的计算如(1)~(4)所示。
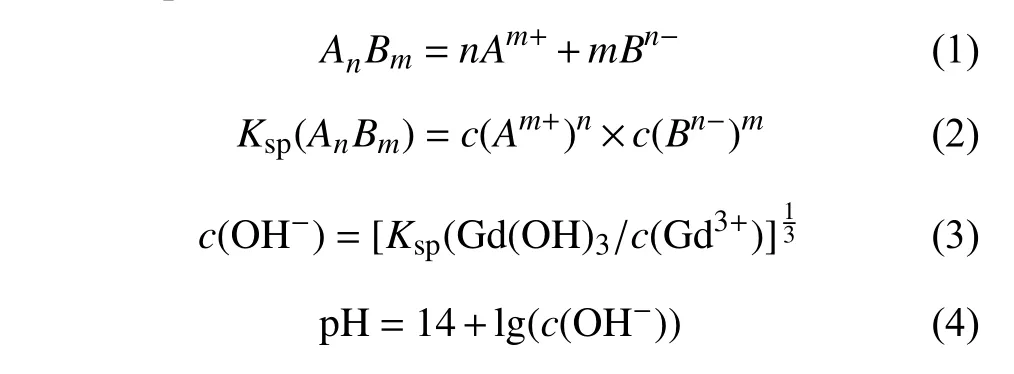
式中:c代表溶解平衡时的物质浓度;Ksp代表难溶物的溶度积。根据稀土氢氧化物的稳定性表可知,Gd(OH)3的溶度积为2.1 × 10–22,由溶度积公式计算可以计算出Gd3+在pH值为6.2以上便开始产生沉淀。本研究以Gd∶Si摩尔比为20∶11配制原料,在反应体系pH值为8~9、9~10、10~11合成了Gd2SiO5前驱体,并在1300 ℃煅烧10 h制备了Gd2SiO5粉体。图4所示为不同反应体系pH值条件下合成Gd2SiO5粉体的XRD图谱。由图4可以看到,在反应体系pH为8~11范围内,所制备粉体的XRD图谱中均出现了高而尖锐的Gd2SiO5衍射特征峰,这说明在该pH范围内合成的Gd2SiO5前驱体均可完全转化成Gd2SiO5。然而,当反应体系pH值为8~9时,所得Gd2SiO5粉体中含有少量的Gd2O3杂质;而当pH值为10~11时,Gd2SiO5粉体则出现了Gd9.33(SiO4)6O2相。对不同反应体系pH值合成Gd2SiO5粉体的XRD分析表明:并流共沉淀法制备Gd2SiO5的过程中,反应体系pH值对粉体材料的物相组成具有明显的影响。
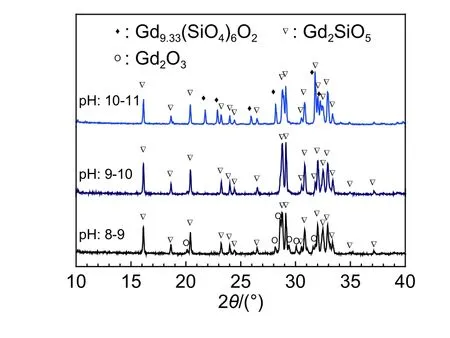
图4 不同反应体系pH值条件下合成Gd2SiO5粉体的XRD图谱Fig. 4 XRD patterns of Gd2SiO5 synthesized with different pH values
在采用并流化学共沉淀法制备Gd2SiO5粉体的过程中,反应体系pH值的升高可使得反应溶液中的OH–浓度增加,增强TEOS水解产物—[Si—O—Si]—网络结构缩聚程度以及Si的活性[31],使得Si更容易被Gd3+所取代而形成—[Si—O—Gd]—网络结构。由前面分析可知,在采用并流共沉淀法合成Gd2SiO5粉体的过程中,原料Gd∶Si摩尔比对所制备粉体材料的物相组成具有明显影响,且合成过程中存在含Si组元的损失。因此,可以推断,当反应体系pH值相对较高时,前驱体中—[Si—O—Gd]—缩聚程度较高,易形成更大的网络结构单元,含Si组元的损失较少,因此煅烧生成的产物中含有少量Gd9.33(SiO4)6O2;而当反应体系pH值偏低时,前驱体中—[Si—O—Gd]—缩聚程度偏低,含Si组元的损失相对较多,从而导致合成的Gd2SiO5粉体中会有Gd2O3存在。Gd2SiO5粉体合成过程中损失的含Si组元可能是相对分子量更小、沉淀结构更加细微的—[Si—O—Si]—悬浮胶体。反应体系pH的升高不仅影响Gd2SiO5粉体材料的物相组成,还可改变沉淀胶体的Zeta电位,增加颗粒之间的排斥力,增加前驱物的分散程度[30]。但是,反应体系pH值过高会延长沉淀洗涤时间,且可促进粒子加速形核,增加颗粒间的碰撞机率,也会导致粉体团聚[32]。因此,反应体系pH值对Gd2SiO5粉体材料的物相组成和分散性均具有重要的影响,反应体系pH值的合理选择对于合成Gd2SiO5至关重要。
2.3 Gd2SiO5粉体的微观形貌
图5所示为Gd∶Si摩尔比为20∶11的前驱体在1000~1200 ℃煅烧10 h制备Gd2SiO5粉体材料的TEM图片。由图5可以看到,在1000~1200 ℃合成温度下,Gd2SiO5粉体的粒径范围在100~200 nm之间,为纳米级粉体。所得Gd2SiO5颗粒形貌呈不规则状特征。另外可以看到,随着合成温度的升高,Gd2SiO5颗粒的平均粒径明显增加。当合成温度较低时(1000 ℃),Gd2SiO5颗粒结晶度偏低,平均粒径较小,且粉体颗粒之间伴随轻微的团聚现象。这是因为Gd2SiO5颗粒粒径很小时,粉体材料具有较大的比表面积,颗粒间的静电吸引力、毛细管力等作用较大,容易产生团聚[33]。而随着煅烧温度的升高,Gd2SiO5颗粒结晶度增加,晶粒尺寸进一步长大,粉体颗粒间可通过烧结而产生团聚。另一方面,高温合成的Gd2SiO5颗粒表面具有相对较高的表面能,纳米颗粒之间通过相互靠拢降低系统自由焓也是导致颗粒团聚的重要原因[34-35]。
为进一步了解Gd2SiO5颗粒的微观形貌特征及单颗粒内部元素分布情况,本研究对在1300 ℃煅烧10 h合成的Gd2SiO5颗粒进行了TEM观察和元素分布表征。图6所示为单一Gd2SiO5颗粒的形貌、高分辨图以及元素分布图。由图6可以看到,该Gd2SiO5颗粒呈现类球形的形貌特征,其颗粒尺寸约为200 nm(图6(a))。图6(b)所示为Gd2SiO5颗粒的高分辨图片。可以看到,Gd2SiO5晶粒内部的晶格条纹清晰,无明显晶格缺陷,晶体中晶面间距(0.31 nm)对应Gd2SiO5晶体中的(021)晶面。图6(c)~(f)所示为Gd2SiO5晶粒内部Gd、O和Si元素的分布图。可以看到,Gd2SiO5晶粒主要由Gd、O、Si三种元素组成,且Gd2SiO5各组成元素在晶粒内部的分布较为均匀。

图5 不同温度合成Gd2SiO5粉体的TEM图 (a)1000 ℃;(b)1100 ℃;(c)1200 ℃Fig. 5 TEM diagram of Gd2SiO5 synthesized at different temperature (a)1000 ℃;(b)1100 ℃;(c)1200 ℃

图6 Gd2SiO5颗粒的微观形貌、高分辨图和元素分布图 (a)微观形貌;(b)高分辨图;(c)GdL分布图;(d)GdM分布图;(e)OK分布图;(f)SiK分布图Fig. 6 Morphology,HRTEM image and element distribution of Gd2SiO5 particle (a)morphology;(b)HRTEM image;(c)GdL;(d)GdM;(e)OK;(f)SiK
2.4 Gd2SiO5粉体的合成机理
采用并流共沉淀法合成Gd2SiO5粉体不同于固相反应法,是一个复杂的物理和化学过程。WU等[22]采用并流共沉淀法合成了Yb2SiO5粉体,并对其合成机理进行了详细的阐述。本研究在前期工作基础上同样对并流共沉淀法制备Gd2SiO5粉体的合成机理进行了探讨。
在本研究中,采用并流共沉淀法合成Gd2SiO5粉体的过程是通过以下几个步骤完成的:首先,Gd2O3通过与HCl反应生成GdCl3溶液,如式(5)所示。同时,TEOS在含有酒精和水的混合溶液中发生水解缩聚生成富含—C2H5和—OH基团的—[Si—O—Si]—网络结构,其形成过程如式(6)所示。

接下来,在将上述两种溶液进行混合搅拌时,TEOS在酸性环境下的水解速率加快,同时Gd3+可被吸附在—[Si—O—Si]—网络结构中形成复杂的结构。而在后续的沉淀过程中,提供了加快水解缩合反应的碱性环境,—[Si—O—Si]—结构中的部分Si便可被临近的Gd3+所替代形成具有网络结构特征的—[Si—O—Gd]—,如式(7)所示。
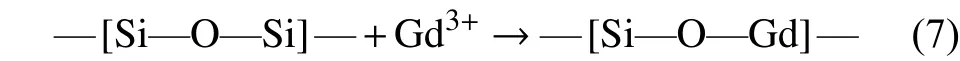
最后在前驱体的烧结过程中,—[Si—O—Gd]—结构经过复杂的物理和化学过程而最终转化成Gd2SiO5(式(8))。在Gd2SiO5前驱体中的—[Si—O—Gd]—网络结构中,当Gd∶Si摩尔比低于Gd2SiO5的化学计量比时,在Gd2SiO5结晶形成的同时,前驱体部分富Si区域会形成Gd9.33(SiO4)6O2相(见公式8);而当前驱体—[Si—O—Gd]—网络结构中的Gd∶Si摩尔比高于Gd2SiO5的化学计量比时,过量的Gd3+便会与氧发生反应生成Gd2O3(见式(9))。
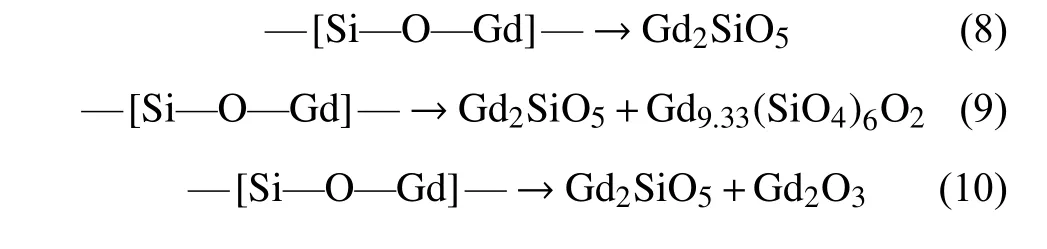
3 结论
(1)共沉淀法合成Gd2SiO5粉体的过程中,前驱体中Gd/Si摩尔比对Gd2SiO5粉体的物相组成具有明显影响。Gd∶Si摩尔比偏低时易生成Gd9.33(SiO4)6O2杂相;而Gd∶Si摩尔比偏高时则易生成Gd2O3杂相。
(2)合成温度的升高对材料的物相组成没有明显影响,但Gd2SiO5晶粒结晶度可得到改善。pH值的变化可明显改变影响—[Si—O—Si]—网络结构缩聚程度以及Si的活性,进而影响Gd2SiO5粉体的物相组成。
(3)Gd2SiO5粉体材料平均粒径为100~200 nm,粉体颗粒呈不规则形貌特征,单一颗粒内部元素分布均匀。
(4)在Gd2SiO5的合成过程中,首先形成具有—[Si—O—Gd]—网络结构特征的前驱体,然后在煅烧过程中逐渐转化为Gd2SiO5晶体以及Gd9.33(SiO4)6O2和Gd2O3杂质相。
