一Stickler综合征家系的临床特征及分子遗传学分析
2021-04-07黄星星刘果高茹心王婷婷范梦杰王希振朱益华吴仁毅张达人刘旭阳
黄星星 刘果 高茹心 王婷婷 范梦杰 王希振 朱益华 吴仁毅张达人 刘旭阳,2
作者单位:1厦门大学附属厦门眼科中心 361000;2深圳市眼科医院 518040
Stickler综合征是一种罕见的遗传性进行性全身性胶原结缔组织疾病,发病率约为1∶10 000,主要是因为胶原蛋白基因突变导致全身广泛胶原蛋白功能紊乱,引起眼部病变,可同时伴有骨骼及口面部发育异常、听力损伤、腭部缺损等一系列变化[1]。该综合征多为常染色体显性遗传,小部分为常染色体隐性遗传,眼部表现较为突出,是儿童孔源性视网膜脱离的常见病因之一[1]。部分患者除眼部表现外还合并严重的骨骼发育异常,而在临床中仅有眼部表现而未合并全身系统病变的患者,常常被漏诊。分子遗传学研究显示75%的Stickler综合征由COL2A1基因突变所致[2]。国外已有较多关于本病分子遗传学的研究报道,中国在该方面的研究鲜见报道。本研究在临床中诊断了1个Stickler综合征的家系,并对其临床表型及基因型进行了分析。
1 对象与方法
1.1 对象
以2019年6月至2020年5月在厦门大学附属厦门眼科中心诊治的一Stickler综合征家系为研究对象。该家系4代共计21人,符合Stickler综合征的临床诊断患者11例,其中男7例,女4例,1例已去世。家系先证者为女性,3 岁。家长代诉自幼视力差,伴听力减退。1%阿托品散瞳验光提示双眼高度近视:右眼-12.00-1.75×75,左眼-11.00-0.75×80。除双眼高度近视外,尚有面部扁平、鼻梁短、关节过度伸展等特征性体征。初步诊断:双眼高度近视,Stickler综合征。为进一步明确病因,本研究对该家系成员进行临床及分子遗传学分析。本研究经所在厦门眼科中心伦理委员会批准(批号:XMYKZX-LW-2019-004),且所有受检者或未成年人的监护人签署知情同意书。
1.2 检查方法
1.2.1 眼部检查 包括裸眼视力、矫正视力、裂隙灯显微镜、间接检眼镜、眼底彩色照相及眼部B型超声等检查。对12岁以下者涂1%阿托品眼膏睫状肌麻痹下验光;12岁及以上者滴复方托品酰胺散瞳验光。
1.2.2 全身检查 包括头面部检查,检查有无腭裂(完全性腭裂、黏膜下腭裂及双叉悬壅垂等),有无Marfan样体型及面中部扁平(颧骨发育不良)、低鼻梁、短鼻、小颌畸形等面部特征。观察受检者面部特征并拍摄正面及侧面照。骨骼及关节检查包括有无关节疼、关节活动度的范围。同时行耳镜及听力检查。家系中已去世成员病史根据病历记录获得。
1.3 分子遗传学分析
1.3.1 遗传学调查及样本采集 对该家系所有成员采用Cyrillic 3.1软件绘制家系图,并进行系谱分析(见图1)。详细记录所有家系成员的发病年龄、全身表现、近视程度、近视度数随年龄的变化及玻璃体视网膜病史。

图1.Stickler综合征家系图示男性患者;示健康男性示健康女性;示女性患者;示先证者;示已故男性;示已故女性Figure 1.Family diagram of Stickler syndrome.represents male patients; represents healthy men; represents healthy women;represents female patients;confirms the proband;represents dead men;represents dead women.
1.3.2 DNA提取 抽取先证者10 ml外周血及家系成员2 ml外周血置于乙二胺四乙酸抗凝管中,按试剂盒说明书方法提取基因组DNA。
1.3.3 全外显子测序 抽提先证者及家系中正常者的基因组DNA,通过琼脂糖凝胶电泳及Nanodrop检测浓度及纯度后进行基因组建库。将样本的基因组DNA随机打断成主峰为200~300 bp左右片段(美国Covaris超声波高性能样品处理系统),使用试剂盒进行末端修复、磷酸化以及加多聚腺苷酸,片段两端分别连接接头制备DNA文库。将文库与带有生物素标记的探针进行液相杂交,使用磁珠将全外显子区域进行捕获并富集后,经PCR线性扩增后进行文库质检,合格后使用美国IlluminaHiSeq 4000测序平台进行高通量测序。
1.3.4 测序数据分析 采用Chromas软件识别基因组单核苷酸变异(Single nueleotide variants,SNV)和插入缺失(Insertion-deletion,InDel)变异;通过与dbSNP(v141)、1000 Genomes等数据库进行比对,过滤掉常见SNV和InDel变异后,筛选得到的变异用于下一步扩大验证。
1.3.5 Sanger测序验证及致病性分析 对全外显子组测序鉴定出的候选变异位点,采用Premier 5.0 设计引物。分别对家系内其他患者及正常人的基因组DNA各个候选变异位点所在的上下游区域进行PCR扩增。将扩增PCR产物纯化后,使用ABL3730XL型DNA测序仪(美国ABL公司)进行Sanger测序,进行家系内基因型-表型共分离分析。应用生物信息软件MutationTaster(http://WWW.mutationtaster.org)对所发现的碱基进行致病性预测分析。
2 结果
2.1 家系特征
该家系4代共计21人,符合Stickler综合征的临床诊断共11 例。其中,男7 例,女4 例。该家系中无近亲通婚,连续4代均有发病者,男女均有发病;凡是患病者,双亲中即有一方患病;若双亲均未患病,则子女均正常,符合常染色体显性遗传模式(见图1)。
该家系患者特征:①高度近视。先证者(IV-3)自幼出现高度近视,右眼眼轴长度(Axis length,AL)27.9 mm,屈光度-12.00-1.75×75;左眼AL 27.2 mm,屈光度-11.00-0.75×80。其余患者也均表现为高度近视。②先天性白内障。家系中包括先证者共5 例患者出现白内障,其中先证者的哥哥(IV-2)右眼裸眼视力(UCVA)0.07,-6.00-3.00×5,矫正视力0.7,左眼UCVA 0.07,-7.00-2.75×175,矫正无助,诊断为左眼先天性白内障,双眼高度近视,双眼弱视,行左眼白内障摘除联合人工晶状体植入术后,矫正视力0.2。先证者的父亲也有白内障。③玻璃体异常。现存10 例患者玻璃体出现不同程度的液化改变。④眼底改变。现存10 例患者中除了1 例患者眼底视网膜脱离外,余患者眼底呈现豹纹状,视盘周围未见明显的萎缩弧(见图2),OCT检查发现先证者的父亲黄斑部形态发育不良,黄斑中心凹厚度增厚。⑤视网膜脱离。家系内有1 例患者出现视网膜脱离,先证者的爷爷(II-1)右眼无光感,左眼0.1,右眼AL16.2 mm,左眼AL30.2 mm;眼部B超提示右眼全视网膜脱离,左眼玻璃体混浊(见图3)。⑥听力障碍。先证者右耳感音性听力障碍,余患者未发现听力障碍。⑦面部特点。家系内现存10 例患者均表现为面中部扁平,低鼻梁、短鼻(见图4)。⑧Marfan样体型。3 例患者四肢较长体型(上部量与下部量比值降低或指距与身高的比值大于1.05),但无明显的鸡胸或漏斗胸(见图4)。⑨关节活动伸展过度。4例患者关节伸展活动度大(见图5)。
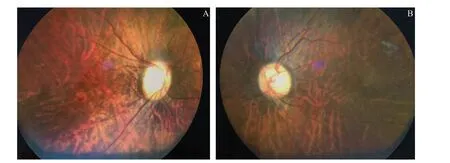
图2.Stickler综合征家系先证者双眼彩色眼底像A:右眼;B:左眼。双眼均显示豹纹状眼底,视盘色淡,边界清晰,视盘周围无高度近视萎缩弧Figure 2.Colored fundus images of both eyes of a proband in a family with Stickler syndrome.A:Right eye;B:Left eye.Leopard-like fundus is shown,optic disc has light color and clear boundary,and there is no high myopia atrophy arc around the optic disc.
2.2 基因检查结果
通过对基因外显子编码区及侧翼区筛查,过滤去掉最小等位基因频率(Minor allele frequency,MAF)>1%的突变多态性位点,比对Clinvar数据库,发现该家系中Stickler患者均有1 个与临床表型共分离的致病性基因变异,COL2A1(NM_033150)基因c.710delG:p.G237fs杂合变异(见图6)。而该家系中正常者未发现该基因突变。该突变导致开放阅读框第710位碱基G缺失,其后部序列发生移码,从原712位后的GTGAAG变为TGAAG,712位翻译密码子TGA为终止密码子UGA,导致其蛋白翻译在第237 位氨基酸残基提前终止,237 位氨基酸残基由甘氨酸G突变为缬氨酸V,且蛋白自第238 位氨基酸残基截断,预测该突变导致蛋白整体结构发生变化从而丧失其正常功能。
3 讨论
3.1 Stickler综合征临床特点
Stickler综合征是遗传性的关节-眼病,患者眼部表现较为突出,较小年龄出现高度近视,与病理性近视不同的是Stickler综合征患者的高度近视为非进展性,眼底视盘周围无高度近视萎缩弧。Stickler综合征患者由于巨大的视网膜裂孔而发生视网膜脱离的风险较高,其中近一半患者可能发生双侧视网膜脱离,主要是与玻璃体先天发育异常有关[3-5]。典型的玻璃体异常表现为“膜型”或“念珠型”,非典型的玻璃体异常表现为不同程度的液化、后脱离或是增殖膜形成,这些变化使得玻璃体后界膜与视网膜分离时裂孔的形成比正常同龄人要早[4]。患者在较小年龄即出现白内障。部分患者因为先天的房角发育异常导致先天性青光眼,但更多的患者是因为视网膜脱离或增殖性玻璃体视网膜病变引起继发性房角关闭[6]。

图3.Stickler综合征家系先证者的爷爷双眼超声检查结果A:右眼视网膜脱离;B:左眼玻璃体内条状混浊Figure 3.Results of binocular ultrasound examination of the grandfather of a proband in a family with Stickler syndrome.A:Retinal detachment in the right eye.B:There is a strip of opacity in the vitreous of the left eye.

图4.Stickler综合征家系先证者面部特征像A:侧面图:低鼻梁、短鼻;B:全身图,Marfan样体型;C:正面图,典型面中部扁平、低鼻梁、短鼻Figure 4.Facial features of the proband of the family with Stickler syndrome.A:Side view:low bridge,short nose.B:Full-body image,Marfan shape.C:Front,typically flat middle,low bridge,short nose.
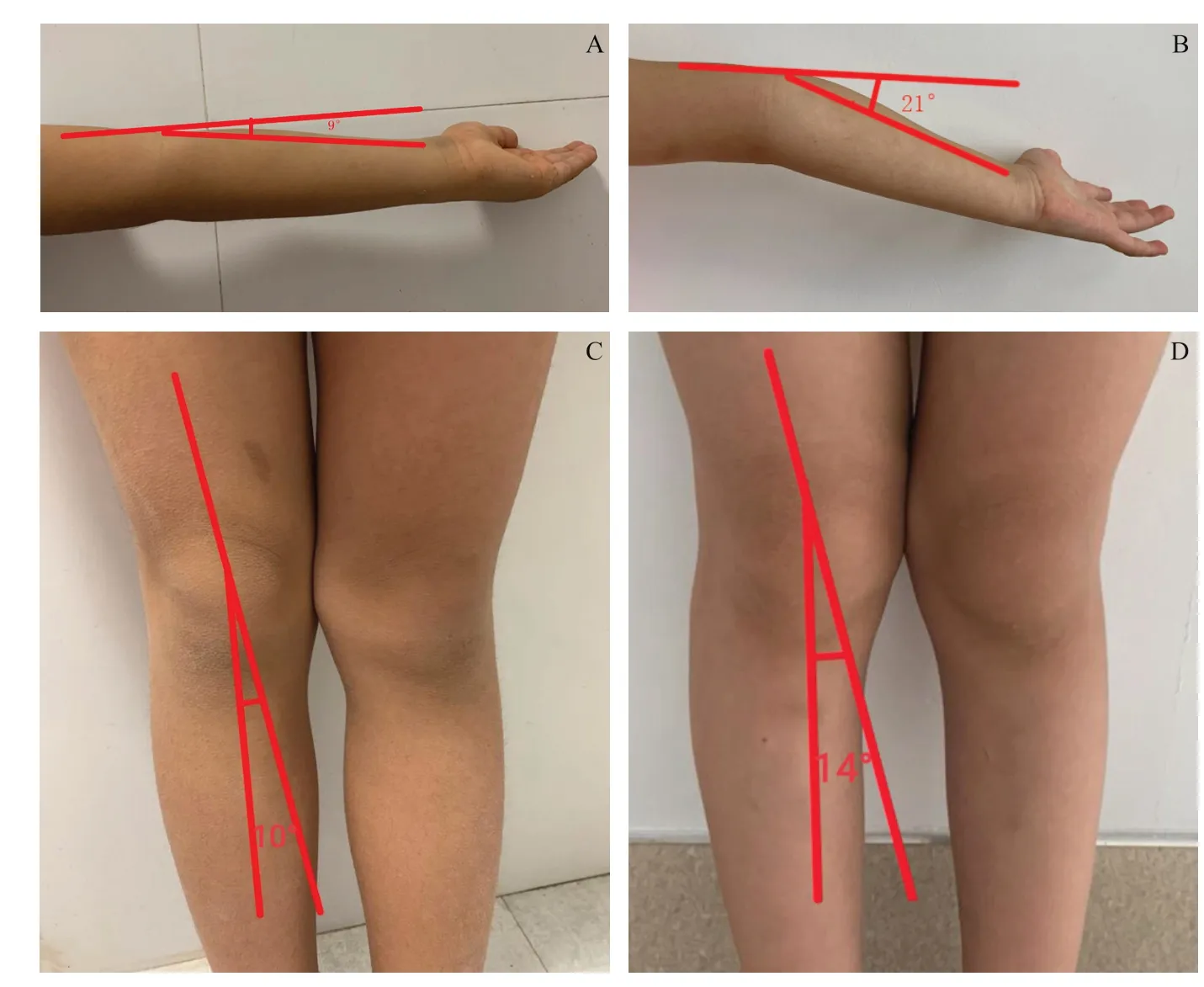
图5.同年龄正常儿童及Stickler综合征家系先证者肘关节及膝关节活动度A:同年龄发育正常儿童肘关节活动度;B:先证者肘关节活动度;C:同龄发育正常儿童膝关节活动度;D:先证者膝关节活动度Figure 5.Range of motion of elbow and knee joint s i n normal age children and Stickler syndrome families.A:The range of motion of elbow joints in children with normal development of the same age.B:The pronator's elbow range of motion.C:Knee range of motion in children with normal development of the same age.D:Range of motion of the knee joint of the proband.

图6.Stickler综合征患者和正常人COL2AI基因序列片段A:杂合的是先证者基因测序片段;B:无变异的是先证者母亲基因测序片段Figure 6.Fragment of the COL2AI gene in patients with Stickler syndrome and normal controls.A:The heterozygosity is the gene sequencing fragment of the probor.B:The one without variation is the gene sequencing fragment of the proband's mother.
Stickler综合征患者听力损伤包括高音区感音神经性听力损伤或内耳及外耳发育异常引起的传导性听力障碍,但其发病机制尚未完全了解,目前比较明确的是在中耳骨之间的关节中存在II型胶原,这些关节内胶原的缺陷可能与传导性听力损伤有关[7]。因为胶原蛋白的功能紊乱,口面部也相应发生了改变,包括腭裂、面中部扁平、低鼻梁、短鼻、小颌畸形等,而腭部异常常与咽鼓管功能受损和中耳功能受损有关。
Stickler综合征的儿童和成人中,超过80%的患者有骨骼发育异常,儿童由于骨骺发育不良表现为骨关节增大、关节活动度过大,可导致平足、关节半脱位或脱位等。其他的骨关节异常还包括脊柱畸形、Marfan样体型,或随年龄增长形成关节病变等,这些骨骼的变化对肢体活动造成了较大的影响。
在对Stickler综合征进行临床评估时需注意。约20%的患者不会发生近视;Stickler综合征有可能发生视网膜巨大裂孔和视网膜脱离的风险,需进行眼底详细检查。听力检查需要区分是传导性还是感音神经性听力损伤。髋关节、腰椎和膝关节的X线检查可显示Stickler综合征的特征。对玻璃体情况评估有困难的新生儿或非常年轻的患者进行临床评估时,可对其父母和兄弟姐妹进行检查帮助诊断。
该家系中所有患者均有眼部玻璃体不同程度液化改变,均有高度近视,年幼时发病,随年龄的增大无明显进展。有1 例发生视网膜脱离,1 例年幼出现白内障,1例自幼听力差。有典型的低鼻梁、短鼻的面容特征,年长者面部体征不明显。家系中患者均有Marfan样体型,先证者及哥哥骨骼关节活动度大,年长者有关节退行性改变。符合Stickler综合征的临床体征。
3.2 Stickler综合征相关的胶原基因及COL2A1基因的相关突变
Stickler综合征相关胶原基因包括COL2A1(COL=胶原;2=II型,A1=α1 肽,以下类推)基因、COL11A1基因、COL11A2基因、COL9A1基因、COL9A2基因及COL9A3基因,分别编码胶原α1(II)、α1(XI)、α2(XI)、α1(IX)、α2(IX)、α3(IX)。COL2A1基因位于12号染色体的长臂12q13.11上,共包含31 538个碱基对,包括54个外显子,第1─5外显子编码Ⅱ型前胶原蛋白的N端,第6─48外显子编码三螺旋结构的核心区,第49 ─52 外显子编码C端。这3 条多肽链折叠成一个杆状的3 股螺旋分子。构成胶原分子3 股螺旋结构的每条链以左手螺旋的方式延长,每圈包括3个氨基酸。每条链都叫做α链[8,9]。这些胶原基因主要决定的是细胞外基质的胶原成分,对于眼部、口面部、关节及听力的发育各自有其独特的作用同时也相互关联。II型胶原(同源三聚体)由COL2A1(COL=胶原;2=II型,A1=α1肽)基因编码,由3条相同的II型胶原多肽链构成。XI型胶原(异三聚体)由3 个不同链组成,每个组成链由3个不同的基因编码:COL11A1、COL11A2和COL2A1。IX型胶原(异三聚体)也是由3 个不同基因编码的3 个不同组成链形成,每个组成链由3个不同的基因编码:COL9A1、COL9A2和COL9A3。XI型胶原的α1链在眼和软骨组织中均有表达,而α2链主要在非眼(软骨)组织中表达。因此,影响后一种蛋白的缺陷会导致关节病,其眼部表型未受影响或正常,而影响前一种蛋白的缺陷会导致眼部和骨骼组织的病变,出现典型的Stickler综合征表型。Stickler综合征患者均存在眼部玻璃体的先天性异常,因为在玻璃体中以II型胶原成分为主,同时也含有XI型和IX型胶原[10]。
COL2A1基因相关的错义突变、缺失、插入、复制及剪切位点突变,大多数是形成提前的终止子的点突变或单个核苷酸缺失导致的移码突变。以往报道过在100例患者的COL2A1基因上找到了77种不同的突变[11,12]。表型变异可能因为:①选择性剪接外显子;②选择性剪接转录的剪接位点突变;③不同氨基酸替代物的差异效应;④镶嵌现象:虽然表面上孤立的COL2A1相关疾病病例可能是由新突变引起的,但有报道强调镶嵌现象是可能的病因[13];⑤杂合变异。Ⅱ型胶原蛋白是关节软骨的主要的结构成分,是稳定的三螺旋结构,该区域核心部分由一段GLY-X-Y重复序列组成,维持关节软骨正常理化性质、力学性能。COL2A1基因的GLYX-Y重复序列中,X位精氨酸向半胱氨酸突变具有显著意义。GLY-X-Y重复序列中的甘氨酸替换可导致多种形式的软骨发育不良,统称为II型软骨病,表现为严重的骨关节病变和躯干短小,可以伴有或不伴有眼部表现。应注意与Stickler综合征鉴别。
本研究通过采用高通量测序技术对眼科遗传疾病相关基因进行全外显子组测序,发现患者的COL2A1(NM_033150)基因c.710delG:p.G237fs杂合变异。在家系中正常人未发现该突变。生物信息学分析示COL2A1基因在黑猩猩、恒河猴、狗、牛、老鼠、老鼠、鸡、斑马鱼和青蛙中均高度保守,197个物种具有人类基因COL2A1的同源基因。Provean预测G237V突变的分值为-7.35,V238del突变分值为-8.31,均为设定阈值2倍以上,均为有害突变。胶原蛋白为重要结构蛋白,此突变致使胶原蛋白链被明显截短,可导致合格胶原蛋白量不足以支撑软骨、眼球有关结构等,使其结构强度降低,诱发相关疾病。
3.3 Stickler综合征的分型
Stickler综合征突变的基因位点不同,因此临床表型及遗传方式差异大,根据是否存在眼部异常、玻璃体表型及分子遗传学特征将Stickler综合征分为5 型[14],I、II及III型的突变基因分别为COL2A1基因、COL11A1基因、COL11A2基因,属于常染色体显性遗传。IV、V型的突变基因分别为COL9A1基因、COL9A2基因,属于常染色体隐性遗传。近年也有关于COL9A3基因突变所致的常染色体隐形遗传疾病。I型Stickler综合征又分为ⅠA型:COL2A1基因外显子2的突变,只有眼部表现,而无全身表现。因为含外显子2转录产物的Ⅱ型胶原蛋白主要存在于玻璃体中,所以ⅠA型只有眼部表现,而无全身临床特征。ⅠB型:COL2A1基因上外显子2以外的突变,临床表现包括眼部表现和全身特征[15]。
综上,本研究通过家系调查、眼科检查及全身相关检查,确定了由COL2A1基因c.710delG:p.G237fs杂合变异的一个ⅠB型Stickler综合征家系,本研究所发现的c.710delG位点的突变在国内外尚未报道。该研究结果丰富了COL2A1基因的突变谱,也有助于遗传咨询和Stickler综合征患者的防治。
利益冲突申明本研究无任何利益冲突
作者贡献声明黄星星:参与选题、设计、家史采集、临床分析、基因测序;进行资料的分析、解释;论文撰写。刘果:参与基因分析,协助论文撰写。高茹心、王婷婷、范梦杰、王希振:协助论文撰写。朱益华、吴仁毅:指导论文撰写。张达人:参与家史采集;协助论文撰写。刘旭阳:参与选题、设计、家史采集、临床分析、基因测序;进行资料的分析、解释;论文修改
