尿黑酸尿症合并周围神经病1例☆
2021-04-06侯文哲詹飞霞蒋青青徐周伟曹立栾兴华
侯文哲 詹飞霞 蒋青青 徐周伟 曹立 栾兴华
尿黑酸尿症(alkaptonuria,AKU)又称为黑尿症或褐黄病,是一种罕见的酪氨酸代谢障碍性疾病,呈常染色体隐性遗传,由编码尿黑酸1,2-二氧化酶的HGD基因(homogentisate 1,2 dioxygenase,HGD)突变所致。患者因先天缺乏尿黑酸氧化酶,导致体内苯丙氨酸和酪氨酸代谢产生的尿黑酸(homogentisic acid,HGA)无法降解,经尿液排出,或慢性进行性沉积于机体多处结缔组织,形成色素沉着。该病早期主要表现为尿色加深、黑尿,皮肤色素沉着,晚期因体内无法排除的尿黑酸侵蚀全身软骨和软组织引起继发性褐黄病性骨关节病[1]。本文报告1例合并周围神经损伤的尿黑酸尿症病例,以增加对该病的认识,防止漏诊、误诊。
1 临床资料
患者,男性,55岁,已婚。因“四肢无力1年,加重2个月”入院。1年前出现四肢乏力,双上肢持物不稳,双下肢借助拐杖缓慢步行数百米,无明显肌肉萎缩、压痛、麻木,无肢体震颤、僵硬等。2个月前感四肢无力较前明显加重,双上肢抬举稍费力,不能下地行走,依赖轮椅出行,伴四肢轻度麻木。为进一步诊治入住我科。发病以来饮食,睡眠及二便无特殊,体重无明显变化。
既往史:10岁发现小便静置后呈棕黑色,尿色深。2012年行腰椎内固定术,2017年行胸椎内固定术,2018至2019年行双侧髋关节置换术。有输血史,无明显输血反应。否认有糖尿病病史。否认家族史,否认父母为近亲婚配。
入院查体:体温36.5℃,脉搏60次/min,呼吸15次/min,血压158 mmHg/75 mmHg,双侧巩膜见片状黑色素沉着,双耳廓见点状黑色素沉着(图1)。神清,语利,查体配合,颅神经无异常。四肢肌张力正常。左上肢近端肌力4级,右上肢近端4级,双上肢远端5级;双下肢近端4级,远端4-级。四肢手套袜套样针刺觉减退,音叉振动觉对称正常。腱反射减弱,双侧病理征阴性,脑膜刺激征阴性。
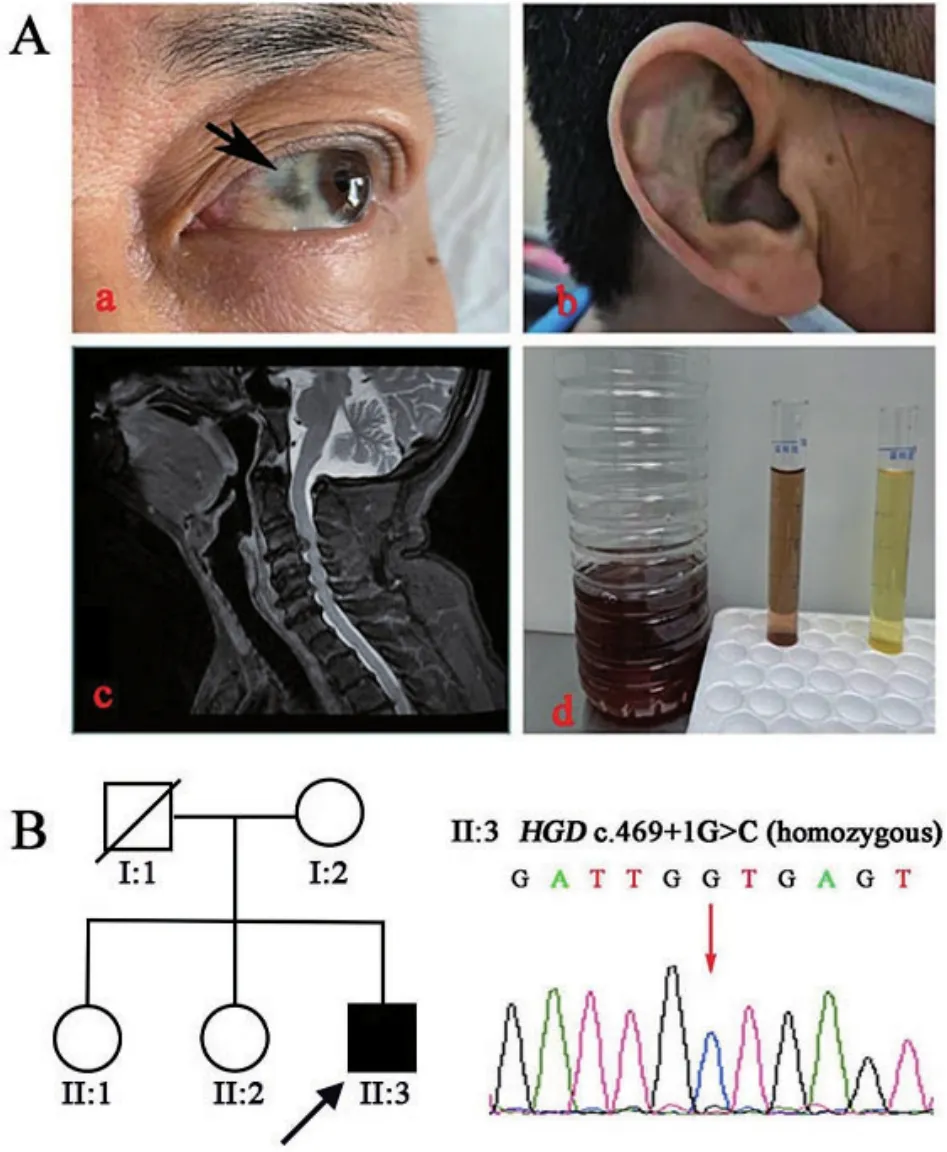
图1 患者的临床表现与基因检测 A.(a)巩膜片状黑色素沉着;(b)耳廓色素沉着;(c)颈椎MRI示C3~C4椎间盘脱出,脊髓受压变性;C4~C5、C5~C6、C6~C7椎间盘突出伴狭窄;(d)尿液静置48 h颜色逐渐加深,呈棕黑色(右1,正常对照)。B.家系图及HGD基因Sanger测序图。
辅助检查:红细胞3.9×1012/L(正常值4.0×1012~5.5×1012/L)、血红蛋白118g/L(正常值120~160g/L);尿素14.0mmol/L(正常值3.2~7.1mmol/L)、肌酐116.9μmol/L(正常值53~106μmol/L)、尿酸458 μmol/L(正常值149~416 μmol/L)、胱抑素C 1.49 mg/L(正常值0.51~1.09 mg/L)。肝肾功能、电解质、血脂、血糖、甲状腺功能、风湿免疫、感染等指标未见异常。颈椎MRI示C3~C4椎间盘脱出,C4~C5、C5~C6、C6~C7椎间盘突出伴狭窄,C3~C4水平脊髓受压变性(图1)。髋关节手术病理报告示滑膜、股骨头关节软骨见黑色素沉着,股骨头髓腔内无菌性坏死,纤维组织增生。肌电图示广泛神经源性损害(颈、胸、腰骶、颅神经段),双侧腓总神经运动神经波幅减低、传导速度减慢,左侧腓浅、右侧尺神经感觉神经波幅减低。
基因检测:患者长期尿色加深,皮肤色素沉着,结合既往多次骨关节手术病史,考虑与尿黑酸尿症相关,首先进行HGD基因一代测序。抽取患者外周血常规提取DNA,采用Primer 3.0软件设计并合成引物,PCR分别扩增该基因的14个外显子及内含子与外显子交界区序列,随后对PCR产物进行Sanger测序。发现患者HGD基因第7号外显子与第8号内含子交界区存在剪切位点纯合突变:IVS7+1G>C(c.469+1G>C)(图1B)。既往文献报告该位点以复合杂合突变形式导致AKU[2],软件预测为致病性。根据“美国大学医学遗传学(American College of Medical Genetics,ACMG)”指南,该变异位点评估为“致病(pathogenic)”[3]。
2 讨论
本文报告了1例合并周围神经损伤的AKU患者,表现为黑尿、巩膜和耳软骨色素沉着、褐黄病性骨关节病,基因检测发现HGD基因IVS7+1G>C(c.469+1G>C)纯合突变。此外,同时合并周围神经损害,扩展了该病的临床表型谱。
AKU是一种极罕见的遗传代谢性疾病,可多系统受累[4]。AKU早期主要为黑尿或尿色加深,色素沉着等,以巩膜和外耳软骨最常见[5]。褐黄病性骨关节病是由沉积的尿黑酸侵蚀全身软骨和软组织所致,多在30岁左右出现[6-7],其早期表现为脊柱、臀部和膝盖处关节疼痛,晚期可出现脊柱、大关节处骨折或肌腱断裂,需行关节置换术或固定术[8]。该病的致病机制除尿黑酸沉积引起过度的氧化应激、炎症反应及细胞毒性之外,也和其他蛋白共同参与有关[9]。尿黑酸可诱导淀粉样蛋白和促炎因子的产生和释放,并伴随氧化应激、淀粉样变性及色素产生[10],淀粉样蛋白沉积的部位同时可见褐黄病色素、组织钙化、脂质氧化、炎性浸润以及氧化损伤和细胞死亡等[10-11]。此外,AKU患者血清淀粉样蛋白显著升高,且在软骨、滑膜、心脏瓣膜、唾液腺等部位出现淀粉样物质沉积[12]。因而目前认为AKU是一种新的继发性淀粉样变性疾病[13]。AKU患者的周围神经损伤及直立位低血压表现,可能是由淀粉样物质在周围神经、自主神经沉积所致[14]。本例AKU患者后续可行腓肠神经活检,进一步观察淀粉样物质、髓鞘和轴索的超微结构。
HGD基因位于3q13.33,由14个外显子组成,编码的尿黑酸1,2-氧化酶(HGD)主要在肝脏和肾脏表达[15]。生理情况下,HGD能将苯丙氨酸和酪氨酸分解代谢过程中的尿黑酸转换为乙酰乙酸,再分解为二氧化碳和水。HGD基因突变导致AKU患者该酶活性减低,尿黑酸代谢障碍而蓄积在体内导致一系列临床症状[16]。自HABBAL等[17]报告第1例AKU后,现已报告200多种突变,热点突变多位于第3、6、7、8、13外显子[18]。国内共报告30余例,尚无确切热点突变[19-21]。HGD突变后导致蛋白亚基稳定性破坏或活性区域残基改变,致使酶活性显著降低[18]。而剪切位点突变可能导致外显子跳跃或从头剪接位点激活[11],该患HGD剪切位点突变可能导致异常结构蛋白翻译,引起酶活性下降。需进一步完善蛋白功能研究,明确该突变的致病机制。
目前AKU尚无有效的治疗方法,以对症支持治疗为主,通过调节饮食限制苯丙氨酸和酪氨酸的摄入,延缓疾病的发生发展[22]。建议大龄儿童和成人每日口服维生素C 1 g,减少尿黑酸在软组织处沉积。药物尼替西农(Nitisinone)能够显著降低AKU患者血浆和尿液中尿黑酸含量[23-25],每天口服10 mg耐受性良好,可有效减少尿中尿黑酸的排泄,减轻褐黄素沉积,延缓疾病进展[24]。副作用包括角膜病变,血细胞减少和卟啉病,因而该药物仍处于临床研究阶段[26-27]。对于大关节受累的患者,早期建议休息、止痛和理疗,晚期进行关节尿黑酸清除术或关节成形术[28]。本病预后尚可,通常不影响寿命,但可因大关节和脊柱受累而致残,死因多为心衰、尿毒症,患者应定期复查心、肾功能[24]。临床高度怀疑该病时,应及早行基因检测有助于早期诊断、治疗及遗传咨询。未来应致力于该病详尽的致病机制研究,以及尿黑酸与淀粉样蛋白等相互作用机理,以改善患者临床症状,提高生活质量。
