苎麻茎皮环状RNA表达谱分析
2021-04-06王延周朱四元刘头明
李 富 王延周 严 理 朱四元 刘头明
专题
苎麻茎皮环状RNA表达谱分析
李 富 王延周 严 理 朱四元 刘头明*
农业农村部麻类生物学与加工重点实验室 / 中国农业科学院麻类研究所, 湖南长沙 410205
苎麻[(L.) Gaud.]是我国特色的天然纤维作物, 其纤维具有拉伸力强、纤维束长等特点。解析苎麻纤维发育调控机制, 对实现纤维产量和品质性状遗传改良具有重要意义。本研究基于高通量测序技术开展了苎麻茎顶部和中部的茎皮组织环状RNA (circRNA)分析, 在2个组织中共鉴定到5268个苎麻circRNA。比较circRNA的表达水平发现, 78个circRNA在2个组织中呈现差异表达。因苎麻茎中部韧皮纤维处于生长发育期, 而茎顶部韧皮纤维尚未起始生长, 推测这78个circRNA是纤维不同发育时期差异表达基因, 它们可能参与了苎麻的纤维发育调控。研究结果将为解析circRNA调控苎麻纤维生长发育机制奠定基础。
苎麻; 纤维发育; 环状RNA; 差异表达
苎麻[(L.) Gaud.]是我国重要的天然纤维作物, 有着悠久的栽培和加工历史, 其纤维及纤维织品是我国重要的工业原料和传统的出口创汇产品。中国作为苎麻的主产国, 其种植面积占全世界90%以上, 因此在国际上苎麻又被称为“中国草”。苎麻纤维是一种韧皮纤维, 具有拉伸力强、纤维长等特点, 其单纤维最长可达55 cm[1], 主要成分为纤维素、半纤维素和木质素。
目前, 苎麻中已有大量可能与纤维发育相关的基因被鉴定, 这为深入解析其纤维形成机制奠定了基础。基于RACE技术, 多个苎麻纤维素合成酶基因()已被鉴定和克隆[2-4]。木质素作为纤维主要成分之一, 其多个生物合成酶基因, 如肉桂酸4-羟化酶()、香豆酸4-香豆酸:辅酶A连接酶()、肉桂酰辅酶a还原酶基因()也被鉴定和克隆[5-7], 并发现它们的表达水平与苎麻纤维木质素含量显著相关[8]。此外, 近10年来高通量测序技术快速发展, 也被广泛应用于苎麻纤维发育机制研究。Liu等[9]分析苎麻转录组共获得36个在韧皮部中高表达的苎麻基因。Chen等[10]比较不同发育时期的苎麻韧皮组织转录组表达谱, 共鉴定到780个差异表达的基因, 其中含4个基因、3个扩张蛋白基因、6个木聚糖水解酶基因, 它们很可能与苎麻的纤维发育相关。
非编码RNA是一类直接发挥催化和调控功能的转录本, 包含miRNA、长链非编码RNA (lncRNA)和环状RNA (circRNA)等[11]。Wang等[12]分析了苎麻纤维在4个不同发育时期的miRNA表达谱, 并在纤维伸长期和次生壁加厚期分别鉴定到150个和148个miRNA, 其中51个miRNA在纤维不同发育期呈现差异表达, 推测非编码RNA可能参与苎麻的纤维发育。circRNA是一类由mRNA前体经反向可变剪切而来的共价闭合且保守的环状转录本, 根据剪接位点可分为exonic circRNAs、intronic circRNAs、exonic-intronic circRNAs和intergenic circRNAs, 它们大多数具有较低的表达量, 且呈现组织特异性和细胞特异性表达。circRNA已知的功能包括隔离miRNA或蛋白质、转录调节和干扰可变剪接, 甚至翻译产生多肽[13]。植物circRNA主要集中于生长发育及生物和非生物逆境研究[14-19], 如Zhang等[14]在玉米和拟南芥中分别鉴定出2174个和1354个干旱相关的circRNA; Zuo等[15]鉴定出番茄163个冷胁迫响应的circRNA; Wang等[16]在野生型和有义/反义转基因番茄果实中发现282个显著差异表达的cicrRNA; Wang等[18]在小麦中发现62个circRNA响应干旱逆境; Gao等[19]在葡萄中鉴定了475个响应寒冷的差异表达circRNA。尽管circRNA被发现广泛参与了植物的生长调控, 但有关它在植物纤维发育中的功能角色, 当前仍鲜有研究。本研究将开展苎麻纤维发育期的全基因组circRNA表达谱分析, 并筛选差异表达的circRNA, 旨在为理解circRNA在植物纤维发育中的调控角色奠定基础。
1 材料与方法
1.1 试验材料和取样
本试验以中苎1号为研究材料。2016年, 将中苎1号扦插苗栽种于中国农业科学院麻类研究所沅江田间试验基地。经切片显微观察, 茎中部韧皮纤维处于生长期, 次生壁正在加厚; 而茎顶部韧皮纤维尚未起始生长[10]。因此, 在2018年5月, 当具有两年龄的苎麻长到约1.5 m高时, 按照Chen等[10]方法, 分别取茎中部(the barks sample collected from the middle, MPS)和顶部(the barks sample collected from the top, TPS)的茎皮组织(图1)。对3株苎麻单独取样, 分别作为3个试验重复。组织样品经液氮速冻后置于-80℃低温保存, 以备后续试验使用。
MPS和TPS分别指从茎中部和顶部收集的茎皮组织样品, 其中位于茎顶端的纤维细胞次生壁尚未加厚, 而处于茎中部的纤维细胞次生壁显著加厚(图片引自Chen等[10])。
MPS and TPS represent the barks sample collected from the middle and top stems, respectively. This picture displays that the secondary cellular walls (SCWs) of fiber cells from the top of the ramie stems do not initiate growth and those from middle part of the ramie stems are thickening. This figure is cited from the study of Chen et al.[10]
1.2 文库构建和高通量测序
利用植物RNA提取试剂TRIzol (Invitrogen, CA, USA)提取6个样品的总RNA。之后对6个RNA样品单独构建circRNA测序文库, 具体流程如下: 使用试剂盒(Ribo-Zero Gold rRNA Removal Kit, Illumina)去除total RNA中的核糖体RNA, 并使用Agilent 2100Bioanalyzer (Agilent Technologies)验证纯化效果; RNaseR反应体系(TruSeq Stranded Total RNA with Ribo-Zero Plant, Illumina, USA)消化线性RNA, 将反应产物纯化, 并打断使其片段化; 以片段化环状RNA为模板, 使用反转录试剂盒(SuperScript III First-Strand Synthesis System for RT-PCR, Invitrogen)先后合成第一链cDNA和第二链cDNA, 并使用试剂盒(PureLink Quick Gel Extraction and PCR Purification Combo Kit, Invitrogen)纯化回收、黏性末端修复、cDNA的3¢末端加上碱基“A”并连接接头, 最后进行PCR扩增; 构建好的文库用Agilent2100 Bioanalyzer (Agilent Technologies)和StepOnePlus Real-time PCR System (ABI)质检, 合格后的文库则通过Illumina测序平台(HiSeq2500, Illumina, USA)进行高通量测序, 从而获得原始的序列reads。
1.3 circRNA预测
对原始序列进行过滤, 主要包括去除接头污染的序列reads、未知碱基N含量大于5%的reads及低质量的序列reads, reads中超过20%的碱基为质量值低于30的碱基(即碱基错误率大于0.032), 从而获得高质量的clean reads。将clean reads比对到苎麻参考基因组上[20], 然后通过CIRI[21]、find_circ[22]2个软件预测circRNA, 并且依据circRNA起始、终止位置来整合2个软件的预测结果。若2个circRNA的起始和终止位置均相近(10个碱基以内), 则将这2个circRNA合并为1个。
1.4 circRNA表达定量及差异表达分析
本研究根据比对至circRNA两端的junction reads数来对circRNA表达定量[23]。因circRNA是由CIRI和find_circ 2个软件预测而来, 故就每个circRNA, 我们计算2个软件注释的junction reads数的平均值, 将其作为该circRNA最终的junction reads数。采用RPB (junction reads per billion mapped reads, 即比对上基因组的所有reads标准化到十亿后跨过backspliced位点的junction reads数目)对各样品作均一化处理。原始的clean reads序列及其表达定量已被提交到DDBJ/EMBL/GenBank数据库(序列号位GSE130587)。
利用DEseq2软件[24]筛选差异表达的circRNA。利用DEseq2软件计算circRNA在茎顶部和茎中部的表达量差异倍数和值, 通过Benjamini-Hochberg统计方法[25]对值进行校正。对于每个circRNA, 若其校正的值小于0.05, 表达差异大于2倍, 则认为该circRNA在2个组织中存在表达差异。
1.5 荧光定量PCR (qRT-PCR)分析
分别提取茎顶部和茎中部RNA (3个生物学重复, 共6个样品), 用DNase I (Fermentas, Canada)进行消化处理, 去除其中的DNA。之后, 利用M-MuLV反转录试剂盒(Fermentas)合成第1链cDNA。使用iTaq Universal SYBR Green super mix试剂盒(Bio-Rad, USA)配置qRT-PCR反应体系, 并在iQ5实时荧光PCR仪上进行PCR。PCR反应程序: 第一步95℃30 s; 第二步40个循环: 95℃15 s, 60 ℃30 s。就每个circRNA, 每个样品进行5个PCR反应重复。18S核糖体RNA在植物各器官中呈现组成性表达, 因此被用作内参基因[12]。circRNA和内参基因引物见表1。依据Livak的2−ΔΔCT方法[26]计算各circRNA在2个组织中的相对表达水平和标准误。

表1 用于qRT-PCR分析的苎麻差异表达circRNA和内参基因引物序列
2 结果与分析
2.1 circRNA的鉴定
经测序, 6个样品共产生大约6.46亿个原始序列reads, 经过滤后得到共计4.45亿个高质量的clean reads, 序列长度合计达到66.7 Gb (表2)。每个样品过滤后均有约74.0百万个clean reads, 且其Q30值均达到96%以上, 说明测序获得的数量和质量均足以开展下一步的circRNA分析。
基于这些高质量的序列, 本研究在苎麻茎顶部和中部韧皮组织中共鉴定到5268个circRNA, 其中539个位于基因间(intergenic), 而4729个(89.8%)则位于基因内部(intragenic)。对这4729个基因进行GO (gene ontology)功能分类发现, 它们主要归类到结合(binding)、催化活性(catalytic activity)、细胞过程(cellular process)、代谢过程(metabolic process)、膜(membrane)等功能类别(Q < 0.01)(图2)。
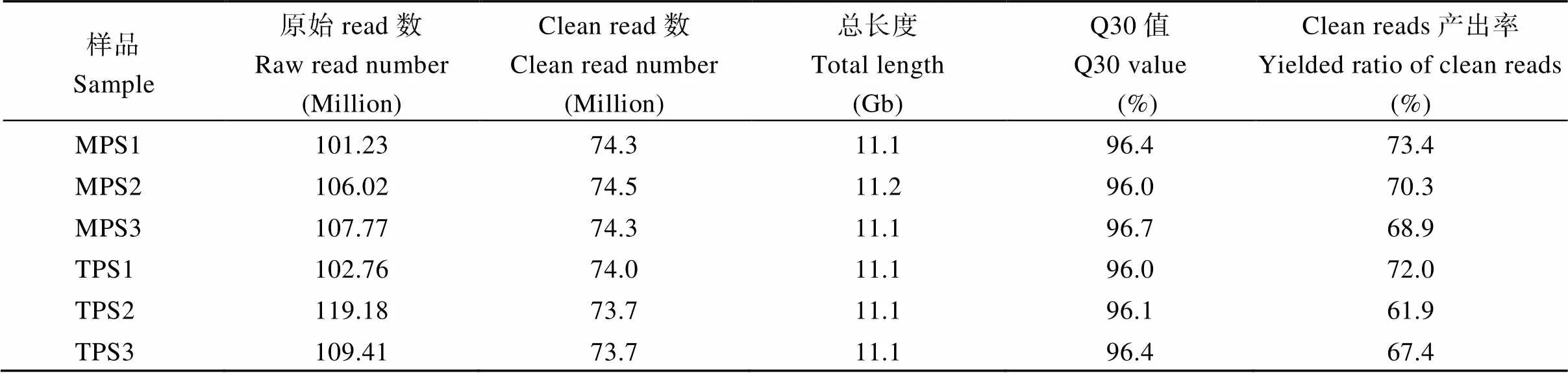
表2 苎麻6个茎皮组织样本的circRNA测序数据统计
MPS和TPS分别指从茎中部和顶部收集的茎皮组织样品; Q30值为序列过滤后的reads中质量值大于30的碱基数占总碱基数的比例。
MPS and TPS represent the barks sample collected from the middle and top stems, respectively; Q30 value indicates the ratio of bases with quality of more than 30 in the clean read.
2.2 circRNA表达谱分析
显微观察显示, 苎麻茎中部韧皮纤维处于生长期, 次生壁正在加厚; 而茎顶部韧皮纤维尚未起始生长(图1)。比较5268个苎麻circRNA在2个茎韧皮部位中的表达水平发现, 相对于TPS, 在MPS韧皮中有77个circRNA显著上调表达, 仅1个circRNA表达水平下调(图3)。因茎中部和顶部的纤维处于不同发育时期, 说明这78个circRNA在2个不同纤维发育时期存在表达差异。在这78个circRNA中, 11个circRNA的表达水平差异倍数达到30倍以上(表3)。其中scaffold13:3497301|3498287和scaffold7:2328081|2328970在MPS中的表达分别上调了42.7倍和42.0倍, 它们分别定位在苎麻基因和中;编码贝壳杉烯酸氧化酶,则为酪蛋白激酶编码基因。此外, 在这78个差异表达的circRNA中, 74个(94.9%)位于基因内部, 其中scaffold1:2974233| 2974487被定位在一个knotted-1-like Homeobox基因()中, 其上调表达倍数达到30.6。
为了验证这些circRNA的差异表达结果, 本研究随机选取了10个circRNA, 通过qRT-PCR进一步分析它们在2个组织中的表达水平。由图4可知, 10个circRNA均呈现出差异表达, 且差异表达倍数明显高于测序分析结果, scaffold6:1851520| 1851960和scaffold30:600099|602644的差异表达倍数甚至达到上千倍。可见, 与高通量测序技术相比, qRT-PCR检测circRNA具有更高的灵敏度。说明这78个circRNA的差异表达是真实可靠的。
3 讨论
纤维广泛存在于植物各器官中, 具有支撑器官形态、植株直立生长, 及抵抗病原菌攻击等作用。此外, 植物纤维也是重要的工业原料, 广泛用于纺织、造纸等工业领域。可见, 不管是对植物本身, 还是对国民经济, 植物纤维具有非常重要的作用。目前, 有关植物纤维生物合成机制在模式植物拟南芥中研究的比较多, 发现其主要由一系列NAC (NAM, ATAF1/2和CUC2)和MYB (v-myb avian myloblastosis viral oncogene homolog)转录因子调控, 它们形成分层次的网络并逐级调控整个纤维发育过程[27]。蛋白翻译后修饰如磷酸化、泛素化等也参与植物纤维的发育调控[28-30]。此外, 非编码RNA也可参与植物的纤维发育调控, 如柑橘可以调控纤维发育控制基因和的表达[31], 而拟南芥则靶向调控基因的表达, 进而抑制纤维发育[32]。
图中红色、蓝色和灰色的数据点分别代表上调表达、下调表达及无表达差异的circRNA; 横坐标和纵坐标分别代表某circRNA在TPS和MPS组织中的read数的对数值。缩写同表2。
Red dots represent transcripts more prevalent in the MPS library, blue dots show those present at a lower frequency in the MPS, and gray dots indicate transcripts that did not change significantly; X and Y axes represent the logarithm of read number for each circRNA in TPS and MPS libraries, respectively. Abbreviations are the same as those given in Table 2.
circRNA是一类重要的非编码RNA, 广泛参与花发育、果实成熟、逆境响应等多个生命过程[33]。植物中存在大量的circRNA, 如PlantcircBase数据库[34]已收录了40,311个水稻() circRNA、38,938个拟南芥() circRNA、6302个玉米() circRNA、7806个大豆() circRNA和1976个番茄() circRNA。Wang等[35]系统研究了244个玉米RNA-Seq样本和288个水稻RNA-Seq样本, 鉴定到38,785个玉米circRNA和63,048个水稻circRNA, 并发现在玉米中有11,206个circRNA响应干旱逆境, 6770个circRNA响应盐逆境, 而在水稻中有824、6313和5724个circRNA分别响应干旱、盐和冷逆境。Zhang等[36]首次发现可能与花生种子发育相关的347个差异表达circRNA, 并通过qRT-PCR验证了其中15个circRNA的表达水平。茉莉酸甲酯(methyl jasmonic acid, MeJA)在植物生长和防御中发挥关键作用, 拟南芥种子用MeJA处理后, 经高通量测序鉴定出8588个circRNA, 其中有385个circRNA响应MeJA处理[37]。已有研究表明, circRNA能够参与调控其宿主基因表达, 例如在水稻中过表达circRNA Os08circ16564降低其宿主基因在叶和花序中的表达[37]。Gao等[19]在低温胁迫下的葡萄叶片中鉴定了475个差异表达的circRNA, 通过异源过度表达一种源自甘油-3-P-酰基转移酶的circRNA (Vv-circATS1), 可以增强拟南芥(Arabidopsis thaliana)的耐寒性, 而源自同一序列的线性RNA则不能, 这为植物耐寒提供新见解。
图4 qRT-PCR分析10个苎麻circRNA在MPS和TPS茎皮中的相对表达水平
Fig. 4 Relative expression level of ten circRNAs stem bark tissues of MPS and TPS by qRT-PCR in ramie
缩写同表2。Abbreviations are the same as those given in Table 2.
当前对circRNA在植物纤维发育中的功能角色仍有待深入研究。本研究鉴定到78个苎麻circRNA在纤维发育不同时期呈现差异表达, 这将为研究circRNA在苎麻纤维发育中的功能奠定重要基础。在78个差异表达circRNA中, 有74个circRNA位于基因内部, 占比达94.9%, 这个比例高于苎麻总circRNA中该类型的比例(89.8%), 其表达是否影响相应基因的表达, 目前仍难以确定。例如knotted-1-like Homeobox家族是植物发育中重要的转录因子家族, 其多个成员, 如拟南芥、棉花均被发现参与植物的纤维发育调控[38-39]。此外, 在纤维发育过程中, 胞外复合体酶EXO70A1主要负责转运纤维素合成酶[40], 而磷酸酶SAC1在纤维次生壁加厚过程中具有重要功能[41]。本研究发现,、和分别注释为拟南芥纤维发育基因、和的同源基因, circRNAscaffold1:2974233|2974487、scaffold23:488065| 489466和scaffold69:125839|129078分别定位到这3个苎麻基因中。因circRNA可通过miRNA海绵功能、干扰可变剪切、结合蛋白等方式调控相应基因表达, 这3个circRNA的上调表达或许会影响到相应的、和表达, 进而可能在纤维发育中发挥功能。因此, 本研究将为我们深入研究这3个circRNA和相应基因的功能及它们的表达调控关系奠定基础。
4 结论
本研究首次在苎麻中鉴定到5268个circRNA, 并发现其中78个circRNA在纤维发育的2个不同时期呈现差异表达。这些circRNA的鉴定为研究苎麻生长发育, 尤其是纤维的生长发育调控功能提供重要基础。
[1] Aldaba V. The structure and development of the cell wall in plants I. Bast fibers ofand., 1927, 14: 16–24.
[2] 蒋杰, 揭雨成, 周清明, 周精华, 朱守晶, 邢虎成, 钟英丽. 苎麻纤维素合酶基因全长cDNA的克隆与表达分析. 植物遗传资源学报, 2012, 13: 851–857. Jiang J, Jie Y C, Zhou Q M, Zhou J H, Zhu S J, Xing H C, Zhong Y L. Full-length cDNA cloning and express analysis ofin ramie., 2012, 13: 851–857 (in Chinese with English abstract).
[3] 刘昱翔, 陈建荣, 彭彦, 黄妤, 赵燕, 黄丽华, 郭清泉, 张学文. 两种苎麻纤维素合酶基因cDNA序列的克隆及表达. 作物学报, 2014, 40: 1925–1935. Liu Y X, Chen J R, Peng Y, Huang Y, Zhao Y, Huang L H, Guo Q Q, Zhang X W. cDNA cloning and expression of two cellulose synthase genes from., 2014, 40: 1925–1935 (in Chinese with English abstract).
[4] 田志坚, 易蓉, 陈建荣, 郭清泉, 张学文. 苎麻纤维素合成酶基因cDNA的克隆及表达分析. 作物学报, 2008, 34: 76–83. Tian Z J, Yi R, Chen J R, Guo Q Q, Zhang X W. Cloning and expression of cellulose synthase gene in ramie [(Linn.) Gaud.]., 2008, 34: 76–83 (in Chinese with English abstract).
[5] 唐映红, 陈建荣, 刘芳, 袁有美, 郭清泉, 昌洪涛. 苎麻肉桂酰辅酶A还原酶基因cDNA序列的克隆与分析. 作物学报, 2015, 41: 1324–1332. Tang Y H, Chen J R, Liu F, Yuan Y M, Guo Q Q, Chang H T. cDNA cloning and analysis of cinnamoyl-CoA reductase gene from., 2015, 41: 1324–1332 (in Chinese with English abstract).
[6] Liu F, Chen J, Tang Y, Chang H, Yuan Y, Guo Q. Isolation and characterization of cinnamate 4-hydroxylase gene from cultivated ramie ()., 2018, 32: 324–331.
[7] Tang Y, Liu F, Mao K, Xing H, Chen J, Guo Q. Cloning and characterization of the key 4-coumarate CoA ligase genes in., 2018, 116: 123–130.
[8] Tang Y, Liu F, Xing H, Mao K, Chen G, Guo Q, Chen J. Correlation analysis of lignin accumulation and expression of key genes involved in lignin biosynthesis of ramie ()., 2019, 10: 389.
[9] Liu T, Zhu S, Tang Q, Chen P, Yu Y, Tang S.assembly and characterization of transcriptome using Illumina paired-end sequencing and identification ofgene in ramie (L. Gaud.)., 2013, 14: 125.
[10] Chen J, Pei Z, Dai L, Wang B, Liu L, An X, Peng D. Transcriptome profiling using pyrosequencing shows genes associated with bast fiber development in ramie (L.)., 2014, 15: 919.
[11] Batista P J, Chang H Y. Long noncoding RNAs: cellular address codes in development and disease.,2013, 152: 1298–1307.
[12] Wang J, Huang J S, Hao X Y, Feng Y P, Cai Y J, Sun L Q. miRNAs expression profile in bast of ramie elongation phase and cell wall thickening and end wall dissolving phase., 2014, 41: 901–907.
[13] Li X, Yang L, Chen L L. The biogenesis, functions, and challenges of circular RNAs., 2018, 71: 428–442.
[14] Zhang P, Fan Y, Sun X, Chen L, Terzaghi W, Bucher E. A large-scale circular RNA profiling reveals universal molecular mechanisms responsive to drought stress in maize and., 2019, 98: 697–713.
[15] Zuo J, Wang Q, Zhu B, Luo Y, Gao L. Deciphering the roles of circRNAs on chilling injury in tomato., 2016, 479: 132–138.
[16] Wang Y, Wang Q, Gao L, Zhu B, Luo Y, Deng Z, Zuo J. Integrative analysis of circRNAs acting as ceRNAs involved in ethylene pathway in tomato., 2017, 16: 311–321.
[17] Yin J, Liu M, Ma D, Wu J, Han B. Identification of circular RNAs and their targets during tomato fruit ripening., 2018, 136: 90–98.
[18] Wang Y, Yang M, Wei S, Qin F, Zhao H, Suo B. Identification of circular RNAs and their targets in leaves ofL. under dehydration stress., 2016, 7: 2024.
[19] Gao Z, Li J, Luo M, Li H, Chen Q, Wang L, Song S, Zhao L, Xu W, Zhang C. Characterization and cloning of grape circular RNAs identified the cold resistance-related Vv-circATS1., 2019, 180: 966–985.
[20] Luan M, Jian J, Chen P, Chen J, Chen J, Gao Q, Gao G, Zhou J H, Chen K, Guang X, Chen J, Zhang Q, Wang X, Fang L, Sun Z, Bai M, Fang X, Zhao S, Xiong H, Yu C, Zhu A. Draft genome sequence of ramie,(L.) Gaudich., 2018, 18: 639–645.
[21] Gao Y, Wang J, Zhao F. CIRI: an efficient and unbiased algorithm forcircular RNA identification., 2015, 16: 4.
[22] Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak S D, Gregersen L H, Munschauer M. Circular RNAs are a large class of animal RNAs with regulatory potency., 2013, 495: 333.
[23] Li L, Guo J, Chen Y, Chang C, Xu C. Comprehensive CircRNA expression profile and selection of key CircRNAs during priming phase of rat liver regeneration., 2017, 18: 80.
[24] Love M I, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2., 2014, 15: 550.
[25] Ferreira J, Zwinderman A. On the Benjamini–Hochberg method., 2006, 34: 1827–1849.
[26] Livak K J, Schmittgen T D. Analysis of relative gene expression data using real-time quantitative PCR and the 2–ΔΔCTmethod., 2001, 25: 402–408.
[27] Zhong R, Ye Z. Secondary cell walls: biosynthesis, patterned deposition and transcriptional regulation., 2015, 56: 195–214.
[28] Speicher T, Li P, Wallace I. Phosphor regulation of the plant cellulose synthase complex and cellulose synthase-like proteins., 2018, 7: 52.
[29] Gui J, Luo L, Zhong Y, Sun J, Umezawa T, Li L. Phosphorylation of LTF1, an MYB transcription factor in populus, acts as a sensory switch regulating lignin biosynthesis in wood cells., 2019, 12: 1325–1337.
[30] Liu C, Yu H, Li L. SUMO modification of LBD30 by SIZ1 regulates secondary cell wall formation in Arabidopsis thaliana., 2019, 15: e1007928.
[31] Huang J H, Qi Y P, Wen S X, Guo P, Chen X M, Chen L S. Illumina microRNA profiles reveal the involvement of miR397a in Citrus adaptation to long-term boron toxicity via modulating secondary cell-wall biosynthesis., 2016, 6: 22900.
[32] Sun X, Wang C, Xiang N, Li X, Yang S, Du J, Yang Y, Yang Y. Activation of secondary cell wall biosynthesis by miR319- targeted TCP4 transcription factor., 2017, 15: 1284–1294.
[33] 骆甲, 王型力, 孙志超, 吴迪, 张玮, 王正加. 植物环状RNA研究进展. 遗传, 2018, 40: 467–477. Luo J, Wang X L, Sun Z C, Wu D, Zhang W, Wang Z J. Progress in circular RNAs of plants., 2018, 40: 467–477 (in Chinese with English abstract).
[34] Chu Q J, Zhang X C, Zhu X T, Liu C, Mao L F, Ye C Y, Zhu Q H, Fan L J. PlantcircBase: a database for plant circular RNAs., 2017, 10: 1126–1128.
[35] Wang K, Wang C, Guo B H, Song K, Shi C H, Jiang X, Wang K Y, Tan Y C, Wang L Q, Wang L, Li J J, Li Y, Cai Y, Zhao H W, Sun X Y. CropCircDB: a comprehensive circular RNA resource for crops in response to abiotic stress., 2019. doi: 10.1093/database/baz053.
[36] Zhang X G, Ma X L, Ning L L, Li Z F, Zhao K K, Li K, He J L, Yin D M. Genome-wide identification of circular RNAs in peanut (L.)., 2019, 20: 653.
[37] Zhang J, Liu R, Zhu Y, Gong J, Yin S, Sun P, Feng H, Wang Q, Zhao S J, Wang Z Y, Li G. Identification and characterization of circRNAs responsive to methyl jasmonate in., 2020, 21: 792.
[38] Li E, Bhargava A, Qiang W, Friedmann M C, Forneris N, Savidge R A, Johnson L, Mansfield S, Ellis B, Douglas C. The Class II KNOX gene KNAT7 negatively regulates secondary wall formation inand is functionally conserved in populus., 2012, 194: 102–115.
[39] Gong S, Huang G, Sun X, Qin L, Li Y, Zhou L, Li X. Cotton, encoding a class II KNOX transcription factor, is involved in regulation of fibre development., 2014, 65: 4133–4147.
[40] Li S, Chen M, Yu D, Ren S, Sun S, Liu L, Liu C M. EXO70A1-mediated vesicle trafficking is critical for tracheary element development in., 2013, 25: 1774–1786.
[41] Zhong R, Burk D H, Nairn C J, Wood-Jones A, Morrison W H, Ye Z H. Mutation of SAC1, anSAC domain phosphoinositide phosphatase, causes alterations in cell morphogenesis, cell wall synthesis, and actin organization., 2005, 17: 1449–1466.
Characterization of the expression profiling of circRNAs in the barks of stems in ramie
LI Fu, WANG Yan-Zhou, YAN Li, ZHU Si-Yuan, and LIU Tou-Ming*
Key Laboratory of Biological and Processing for Bast Fiber Crops, Ministry of Agriculture and Rural Affairs / Institute of Bast Fiber Crops, Chinese Academy of Agricultural Sciences, Changsha 410205, Hunan, China
Ramie [(L.) Gaud.] is a special natural fiber crops in China, and its fiber has many excellent characteristics, including long strands and well tensile strength. Elucidation of the mechanism for fiber formation will be helpful for the improvement of fiber yield and quality in ramie. In this study, the expressed analysis of circular RNA (circRNA) for the tissues of barks from the top stems and middle stems were performed by Illumina sequencing, respectively. The total of 5268 circRNAs were identified. Among these circRNAs, 78 showed differential expression between two examined tissues. Previous cytological observation suggested that the secondary cellular walls (SCWs) of fiber cells from the top of the ramie stems did not initiate growth and those from middle part of the ramie stems were thickening. Therefore, we speculated that these 78 differentially expressed circRNAs were potentially involved in the fiber development in ramie. The results provide an important basis for understanding the role of circRNA in the regulation of fiber development.
ramie; fiber development; circRNA; differential expression
10.3724/SP.J.1006.2021.04042
本研究由国家自然科学基金项目(31871678)和中国农业科学院科技创新工程项目(CAAS-ASTIP-IBFC)资助。
This study was supported by the National Natural Science Foundation of China (31871678) and the Agricultural Science and Technology Innovation Program of China (CAAS-ASTIP-IBFC).
刘头明, E-mail: liutouming@caas.cn
E-mail: 704510340@qq.com
2020-02-24;
2020-07-02;
2020-07-16.
URL: https://kns.cnki.net/kcms/detail/11.1809.S.20200715.1817.006.html
