微囊型脂肪粉中粗脂肪的快速测定
2021-03-26殷秋妙王威利黄晓梅孙丽华
殷秋妙,王 勇,强 莉,王威利,黄晓梅,孙丽华
(1广东省农业科学院农业质量标准与监测技术研究所,广州510640;2农业部农产品质量安全检测与评价重点实验室(广州),广州510640;3广州市优百特饲料科技有限公司,广州510550;4山东省饲料质量检验所,济南250022)
0 引言
油脂是动物全价饲料的重要原料之一,在降低动物热应激、满足特殊生理期能量需要、为动物提供必须脂肪酸、提高免疫力等方面具有重要生理作用[1-3]。饲料用油中,传统的原料油在脂肪酸组成上不平衡,较难被动物消化吸收,且容易氧化酸败进而带来了养殖风险[4-6]。针对这些问题,近十多年来,国内外企业利用微囊化技术[7]创造了新型饲料原料——微囊型脂肪粉。微囊型脂肪粉是以油脂为芯材,以天然碳水化合物和(或)蛋白质等高分子生物可降解物质为壁材,经物理化学工艺(目前主要是喷雾干燥法)处理而成为“水包油”型微小脂肪粉[8-9],粒径为5~500µm,脂肪含量多在50%以上。微囊型脂肪粉已在仔猪、繁育母猪等动物的养殖业中得到了较广泛的应用。
目前,食品和饲料中粗脂肪测定主要有索氏抽提法[10-12]、碱水解法[10-13]、氯仿-甲醇法[14-15]和酸水解法[10-12]。由于油脂被包裹,索氏抽提法测定结果偏低[14];氯仿-甲醇超声破解法可用,但氯仿试剂毒性较大,不适宜标准化;碱水解法在乳制品中应用较多,但不适于水解微囊型脂肪粉的壁材大分子物质;酸水解法理论上可行,但现有的2个国家标准[10-11]中酸水解方法对微囊型脂肪粉不适合,测定结果均偏低,偏低原因主要是微囊型脂肪粉粗脂肪含量高而水相转移造成损失过大[10],和水解温度偏低造成水解不完全引起[11],并且GB/T6433—2006[10]的方法步骤非常繁琐、耗时。国外也尚未公开这类产品的测定方法,因而这将影响该产业的发展和产品贸易。
基于此,本研究按照试验方法标准的编写规则[16]要求,以在全国范围内广泛收集的微囊性脂肪粉代表性样品(标准化的对象)为材料,根据酸水解原理,通过筛选合适的酸水解条件及萃取方法,首次建立了微囊型脂肪粉中的粗脂肪含量的快速测定方法,并依据GB/T 6379.2—2004测量方法与结果的准确度(正确度和精密度)[17]的要求,组织了实验室验证,确定了该方法的性能指标,为该方法的标准化确定了适用范围和主要技术步骤及指标等规范性技术要素。
1 材料与方法
1.1 主要仪器
FOSS SoxtexTM8000脂肪仪(瑞典);DHG-9240A电热恒温鼓风干燥箱(上海一恒);HH-4数显恒温水浴锅(常州澳华);Eppendorf 5804R台式高速离心机。
1.2 试剂与材料
盐酸、石油醚、95%乙醇均为分析纯(广州化学试剂厂);代表性样品由广州市优百特饲料科技有限公司收集提供,是从国内大型生产厂家收集到的,覆盖了国内该类产品生产厂家的80%,共5份微囊型脂肪粉样品和4份载体脂肪粉样品。FA-6样品是某大型企业检验新方法的试验样品。
1.3 实验方法
样品称取1~2 g(精确到0.1 mg),放入50 mL离心管中,加入盐酸溶液20 mL,在水浴锅中水解一定时间,取出置室温稍冷,加石油醚20 mL振荡2 min,5000 r/min离心5 min,吸取上层液(石油醚层)于已经恒重铝筒(103℃±2℃,0.5 h,干燥器中冷却)中,重复萃取,石油醚萃取液用脂肪仪去溶剂后,在103℃烘箱干燥15 min后,取出置干燥器中冷却后称重,残渣重即为粗脂肪的质量。盐酸浓度、水浴温度、水解时长、萃取次数等重要技术参数通过筛选确定。
试样中(粗)脂肪含量的计算见公式(1)。

式中:W—试样中(粗)脂肪的含量,%;A—样品的质量,g;B—铝筒的质量,g;C—铝筒和(粗)脂肪的质量,g。
表面油的测定:按照GB/T 6433中A类样品方法测定。
包埋率的计算见公式(2)[13,15]。

1.4 方法验证
添加回收试验:以编号为FA-6的微囊型脂肪粉为试材,称取4份样品,每份1 g,添加2个梯度,添加量分别为0.2 g和0.4 g大豆油,依据所建方法测定粗脂肪;计算方法的重复性(以标准差表示)和回收率。
实验室间比对[17]:委托3家实验室对5个包埋率水平不同的微囊型脂肪粉样品,每个样品重复测定6次,依据测量方法与结果的准确度标准,对所得数据进行柯克伦(Cochran)检验,以评价该方法在实验室间精密度的一致性;用格拉布斯(Grubbs)检验判定实验室间平均值的一致性,若一致则判定结果准确;根据再现性方差=重复性方差+室间方差,即:SR2=Sr2+SL2,计算再现性标准差,通常把2.8倍作为方法允许误差系数,即LSr=2.8*Sr,LSR=2.8*SR,并依此推算确定方法的重复性和再现性允许差。
2 结果与分析
2.1 方法的建立
2.1.1 试验样品包埋率测定 为使建立的粗脂肪测定方法具有良好的适用性,宜选用包埋率较高和较低两端位的微囊型脂肪粉作为试验材料。为此,用GB/T 6433—2006[10]中索氏抽提方法对收集到的微囊性脂肪粉样品的表面油含量进行了测定,而总油含量以企业给出的产品标示值计,进而了解样品油脂包埋率情况(表1)。从表1可以看出,5份样品的脂肪包埋率分布宽泛,差异较为悬殊,代表性较好;样品FA-4的脂肪包埋率最高,达83.40%,样品FA-5的脂肪包埋率最低,为18.83%。本研究选择脂肪包埋率最高的FA-4样品和脂肪包埋率最低的FA-5样品作为试验材料进行方法的建立研究。
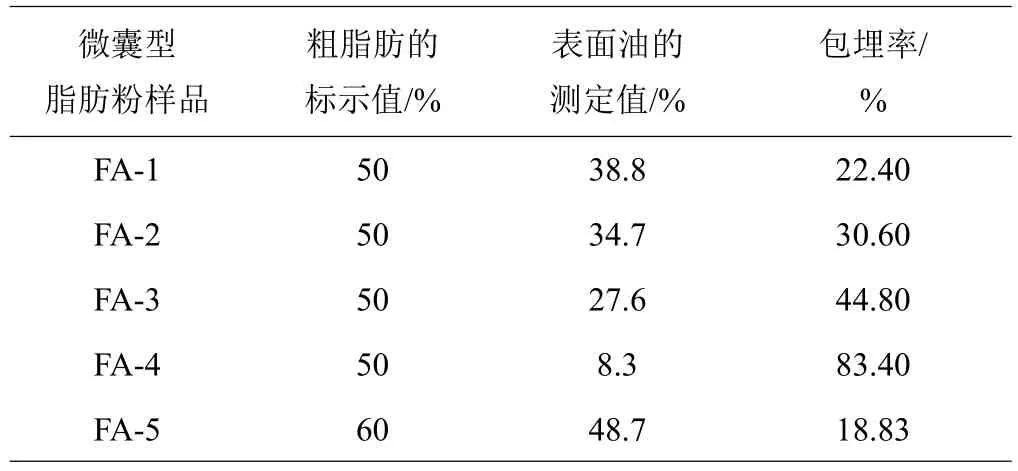
表1 5种微囊型脂肪粉包埋率的测定
2.1.2 酸水解条件筛选 酸水解的设计涵盖现有国家标准[10-11]中酸水解的条件并扩大了试验范围,在水解时间为60 min时,研究了4种盐酸浓度(3 mol/L、4 mol/L、5 mol/L和6 mol/L)和4种水解温度(70℃、80℃、90℃和100℃)对包埋率最高的FA-4样品和包埋率最低的FA-5样品中粗脂肪含量测定的影响。考虑到微囊型脂肪粉的壁材特点,如蛋白类壁材多用乳清蛋白,而乳清蛋白的热凝胶变性的温度在75℃左右[18],以及多糖类壁材比蛋白类更容易水解,因此水解试验温度以70℃为起点。
实验过程中发现,由于微囊粉的亲水性不强,容易漂浮在酸溶液表面,水解酸度的增加虽然能够加快水解反应,但同时带来了加速美拉德褐变和使脂肪氧化的风险。6 mol/L盐酸浓度的水解条件均可将2种实验样品的微囊壁完全地水解,但溶液完全褐变;此外,试验操作过程中因酸度过高也有一定的安全风险,因而不宜选用。
其他组合条件下测定结果见图1和图2,可以发现,水解温度80℃和90℃的两个处理组因酸度引起的2个样品中的粗脂肪含量测定结果变异较小,均十分接近真实值;70℃和100℃处理组酸度变化引起的测定结果变异较大,且100℃时包埋率高的样品可能存在未加抗氧化剂因氧化作用引起的整体偏高特点。在80℃和90℃的水解温度条件下,3 mol/L和4 mol/L盐酸水解时粗脂肪的测定结果一致性更好。鉴于饲料市场中目前及未来可能有包埋率更高的微囊型脂肪粉产品,以及出于水解充分和减少褐变的考虑,选择用4 mol/L盐酸85℃±5℃水解1 h作为微囊型脂肪粉的酸水解条件。

图1 不同水解条件对微囊型脂肪粉FA-4样品中粗脂肪含量测定的影响

图2 不同水解条件对微囊型脂肪粉FA-5样品中粗脂肪含量测定的影响
水解时间能否缩短以建立更加快速的测定技术呢?笔者以包埋率最高的FA-4样品为材料,设计了水解时间为30 min到1 h内的处理进行了研究,以该温度下样品溶液出现褐变为该温度的水解终止点,具体为 70℃水解 60min、80℃水解 40 min和 90℃水解30 min。从结果(图3)可以看出,在70℃水解60 min条件下,不同盐酸浓度处理的粗脂肪测定结果基本一致,相对比较接近真实值;在80℃水解40 min和90℃水解30 min条件下,随着盐酸浓度由低到高,粗脂肪的测定结果均呈现出逐渐增大的规律,但测定结果仍然偏低,只有在盐酸浓度为6mol/L时测定结果才比较接近产品的真实值。这说明,在盐酸浓度从3mol/L增加到5mol/L的情况下,即使水解温度提高,水解时间从60 min缩短到40 min乃至30 min造成了微囊壁的水解不完全,进而造成了测定结果偏低。因此,水解时间还是以60 min为好。
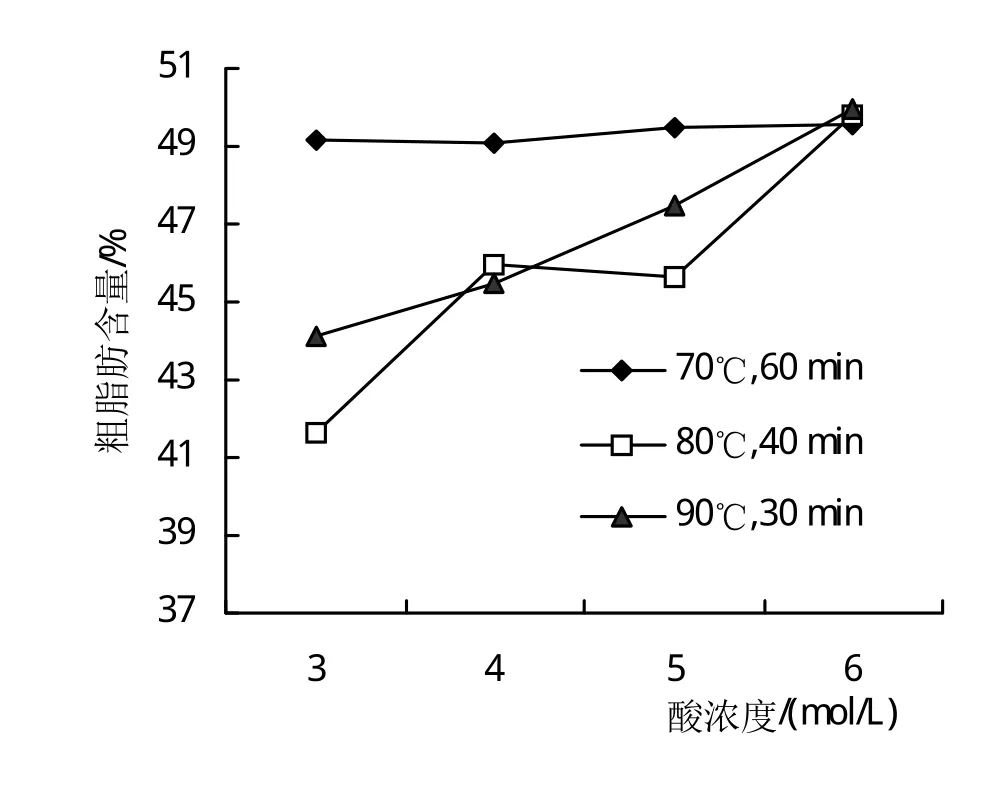
图3 不同水解条件对包埋率较高的FA-4样品粗脂肪含量测定的影响
2.1.3 萃取方法的选择 在食品脂肪测定[11]方法里,采用无水乙醚或石油醚作为酸水解后萃取剂。邱清莲等[19]比较了乙醚和石油醚萃取对酸水解含糖样品中的粗脂肪测定的影响,发现用石油醚测定脂肪的准确度高,而用乙醚萃取的粗脂肪测定值偏高,准确度下降。这是由于乙醚具有一定极性,能够萃取部分碳水化合物,而石油醚不能[20]。鉴于多糖类是部分复合微囊壁材的重要成份之一,为了避免糖类对方法准确性的影响,采用石油醚作为萃取剂。
萃取时溶剂乳化和粘附在石油醚层容器壁上残渣也将影响测量结果的准确性。这些残渣是带有非极性氨基酸的球蛋白残片,通过加入乙醇,使乙醇的亲油端与残片结合使其迅即沉入下层酸水溶液中,进而去除;此外,乙醇还具有降低乳化作用,再辅助以离心很好地解决了乳化和残渣影响。
以FA-5样品为试材,称样2 g,加3 mol/L和4 mol/L盐酸溶液20 mL,85℃水浴1 h,期间每20 min涡旋15 s,冷却至室温,用20 mL石油醚萃取,加入95%乙醇5 mL消除黏附在石油醚层器壁上的残渣和去乳化,蒸馏除去溶剂(可用脂肪仪去溶剂)后,于103℃烘干;重复用20 mL石油醚萃取,分析各次萃取脂肪的比率,从而确定萃取次数。从结果(表2)可以看出,在2种浓度的盐酸水解处理中,第1次石油醚的萃取率均高于90%,2次石油醚的萃取率已高于99.7%,经过3次萃取后萃取率均达100%。

表2 石油醚萃取次数对微囊型脂肪粉FA-5中粗脂肪含量测定的影响
从FA-5样品的萃取试验的结果中发现,40 mL石油醚至少可以萃取2.2 g脂肪。本研究中所用实际样品粗脂肪含量在50%~60%,文献报道有含量为64%[21]。对于本方法建议的称样量1~2 g,2 g的称样量,即使粗脂肪含量100%,也即为2 g,40 mL石油醚的萃取依然是充足的。
综上所述,着眼于绿色环保和满足分析精度要求的理念,萃取方法选择石油醚萃取2次,每次20 mL。此外,还对去溶剂后粗脂肪残渣的烘干时间进行了研究。烘干的目的是完全去除粗脂肪中的有机溶剂,确保粗脂肪的测定值准确。试验发现,15 min烘干和30 min烘干后粗脂肪的重量一样(结果未示),这说明烘干15min就可以将粗脂肪中的有机溶剂全部蒸发出去,这一结果同GB/T6433—2006[10]中的烘干时间一致。
2.2 方法性能考察
2.2.1 室内重复性和正确度 添加回收试验结果详见表3,FA-6样品粗脂肪平均含量为52.72%,RSD为0.08%,大豆油的平均回收率为100.0%,RSD为1.1%。对照GB/T 27404—2008实验室质量控制规范[22]中对测定结果的精密度和回收率的要求(含量在10%~100%时,室内精密度要求1.3%≤RSD≤2.0%,回收率要求为95%~105%[22]),结果表明本研究的测定结果符合GB/T 27404—2008的要求。

表3 所建方法的添加回收试验
2.2.2 实验室间比对和方法精密度确定 3家实验室测定平均值和室内平均标准差见表4,各实验室测定结果的标准差经过柯克伦(Cochran)检验[17]结果无离群值,平均值经格拉布斯(Grubbs)检验[17]无离群值,说明测定结果准确。5个样品室内重复性误差(平均标准差)为0.18%~0.35%,平均为0.27%;再现性误差(标准差计)为0.20%~0.72%,平均为0.46%;依据重复性误差和重复性误差限的关系,推算出方法的重复性限0.75%,再现性限为1.3%。因此,本新建方法的允许误差宜表述为“在重复性条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的1.5%。

表4 所建方法的实验室结果比对和方法精密度确定
2.3 所建方法与现行标准GB/T6433—2006的比较
用所建方法和GB/T 6433—2006中酸水解方法测定FA-4和FA-5样品粗脂肪含量(表5),并按照GB/T 6433—2006中的方法计算精密度。结果表明,所建方法的粗脂肪测定结果与产品的真实值更吻合,而GB/T 6433—2006中酸水解方法的测定值明显偏低;所建方法的精密度明显优于国标GB/T 6433—2006方法。测定脂肪包埋率较高的样品FA-4和包埋率较低的样品FA-5时,国标GB/T 6433—2006方法的测定值分别比所建方法低29.1%和33.3%。
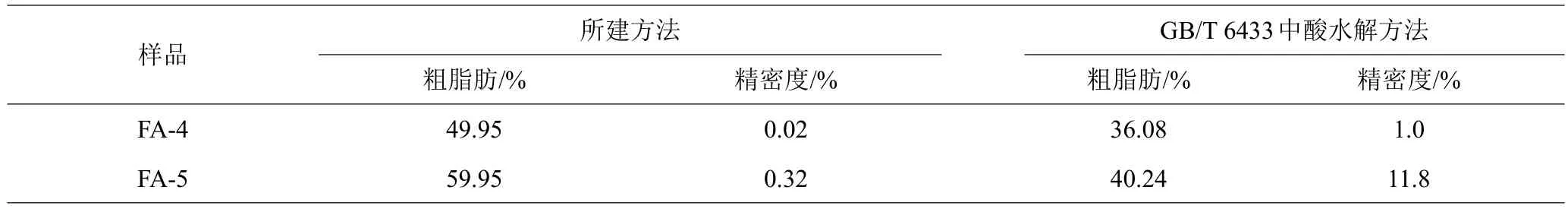
表5 所建方法与GB/T 6433酸水解方法测定微囊型脂肪粉粗脂肪结果的比较
本研究所建方法操作简便,且完成测定所需要的时间短,约为2.6 h,而国标GB/T 6433—2006[10]酸水解法约需要7 h;此外,新建方法较为经济和环保,节约了大量有机试剂的使用。
2.4 所建方法测量载体脂肪粉中粗脂肪含量的结果分析
将所建方法应用于5种载体脂肪粉样品测定,并与索氏抽提方法比较,结果见表6。所建方法比索氏抽提法测定值偏低,低24%~10%,造成此结果的原因在于2种方法提取机理的差别。本研究所建的酸水解萃取法对粗脂肪的选择取决于2个方面,即在石油醚中的溶解度以及在石油醚和酸溶液中的油水分配系数,而索氏抽提法对粗脂肪的提取仅指在石油醚中溶解的物质。载体脂肪粉是由膨化玉米等作为载体物理吸附油脂(包括磷脂),油脂处于游离状态,容易被石油醚抽提,不需要酸水解。而酸水解过程载体脂肪粉样品中的磷脂在强酸加热条件下可完全水解成甘油、脂肪酸、磷酸等,磷酸溶解于水,因而致使结果偏低。因此,本研究的所建方法不适合载体脂肪粉样品中粗脂肪含量的测定。

表6 2种方法测定载体脂肪粉结果比较
3 讨论与结论
食品工业把微囊型脂肪粉以粉末油脂命名[14],饲料工业还没有标准化的命名。由于饲料中也有粉末化的载体脂肪粉,为突出微囊型脂肪粉产品的技术特点,又兼顾到其与饲料工业标准化术语[23]“微胶囊化”的异同,本课题组将其命名为微囊型脂肪粉。目前在现行食品和饲料粗脂肪检测标准中均未涉及该类产品。本研究首先尝试了食品和饲料的相关方法[10-11],发现均不适用;其他方法中选用的试剂毒性较大,不符合标准化的普适、环保等要求。
影响微囊型脂肪粉粗脂肪测定准确度的关键有3点,首先是微囊壁是否全部破碎,其次是萃取时溶剂乳化和粘附在石油醚层容器壁上残渣造成的干扰,第三是样品不含磷脂。本研究没有直接研究破坏微囊壁结构的物理化学过程,而是从微囊破壁的条件以及与之密切相关的游离脂肪的质量入手,确定水解条件。4 mol/L盐酸85℃下水解1 h,足可以使所有热凝胶蛋白二级以上结构破坏,多糖类壁材比蛋白更容易水解,因此无论对于单材料(多糖类或蛋白类壁材)或二者组合的复合壁材,水解条件均适用。水解过程不同样品发生褐变程度不一,褐变主要是糖和蛋白类氧化发生美拉德反应引起,脂肪在此环节可能会微量氧化,但是试验证明这些并没有引起粗脂肪质量的明显变化。在方法最后去溶剂烘干环节,脂肪应该会发生轻微氧化,但是和油脂氧化稳定性测定所需要的60℃30天加速氧化法相比,氧化问题可以忽略不计,这也是所有脂肪测定方法[10-12]所默许的。乙醇的使用很好解决了黏壁和乳化问题,此效应同文献[11]一致。目前,饲用磷脂常以磷脂粉或者载体脂肪粉形式应用。磷脂具有乳化和亲水性,容易水解和氧化。在药物微囊化技术中的脂质体技术中有使用,但是因脂质体的热稳定性和化学稳定性较差,该技术目前还不成熟[7],而且生产工艺和成本高,因此饲用微囊型脂肪粉生产中不会添加磷脂。本研究的创新点是建立了新的酸水解方法,使之适用于微囊型脂肪粉,并优化了萃取方法,使之充足又避免了浪费,实现了方法的精准高效和经济。
此外,本研究过程还发现微囊型脂肪粉另一个质量指标——包埋率仍然缺乏测定方法标准。包埋率由表面油和总油推算而来。目前表面油测定方法不一;本研究用索氏抽提法测定表面油,比其他文献报道[13,15]的抽提时间长,包埋率相对数值会低一点,但这样做可以更好地了解样品的多样性和包埋率。此法测得的测试样品包埋率19%~83%,样品的代表性比较好。
本研究所建方法为:称取样品1~2 g(精确到0.1 mg)于50 mL离心管中,加4 mol/L盐酸20 mL,拧紧盖子,涡旋30 s,85℃±5℃水浴1 h,期间每20 min涡旋15 s,水解结束,取出置冷却至室温,开盖加20 mL石油醚和5 mL 95%乙醇,拧紧盖子,用力振荡40 s,室温下,5000 r/min离心6 min,吸取上层液于已经烘干恒重过的铝筒中,再加入20 mL石油醚萃取,合并2次上清液于铝筒中,蒸馏除去溶剂(可用脂肪仪去溶剂)后,于103℃±2℃烘干15 min,在干燥器中冷却称重。本方法适用于不含磷脂的微囊型脂肪粉中粗脂肪的测定,方法重复性误差(平均标准差)为0.18%~0.35%,平均为0.27%;再现性误差(标准差计)为0.20%~0.72%,平均为0.46%;方法的重复性限0.75%,再现性限为1.3%。方法各项性能指标满足实验室质量控制规范[22]指标要求,比现行标准方法[10]节省试剂50%以上,节约时间50%以上,方法快捷、经济,已经被相关企业采用。
