铁死亡调控机制及在肺癌治疗中的研究进展
2021-03-25陈维达郭溟浩陈泽涛
徐 飞,陈维达,郭溟浩,陈泽涛
(山东中医药大学附属医院 老年医学科,山东 济南 250014)
肺癌(lung cancer,LC)是全球肿瘤相关死亡的主要原因之一,其高发病率、高病死率成为世界关注的焦点。然而,传统的治疗手段以及分子靶向药物、肿瘤免疫疗法并没有为晚期或复发患者带来理想的利益。面对上述治疗困境,进一步探究肺癌的发生发展机制,发现新的治疗靶点,寻找有效的治疗方法与药物,是肺癌研究领域亟需解决的问题。
既往根据细胞形态,将细胞死亡分为3种类型:凋亡(apoptosis)、自噬(autophagy)和细胞坏死(necrosis)。2012年,一种铁依赖性的脂质过氧化损伤导致的新型非凋亡细胞死亡模式被提出,命名为“铁死亡” (ferroptosis)[1]。它本质上是由膜脂修复酶——谷胱甘肽过氧化物酶(glutathione peroxidase 4,GPX4)活性失效、细胞内脂质过氧化物代谢障碍、铁依赖的脂质活性氧自由基(reactive oxygen species,ROS)大量累积所致的细胞死亡,在形态学、遗传学、生化特征上与凋亡、坏死、自噬具有显著差异[1]。在形态学上,铁死亡主要表现为线粒体体积缩小、线粒体膜密度增加、线粒体嵴减少甚至消失、外膜破裂,而无细胞核浓缩、染色质边缘化[1]。与正常细胞相比,肿瘤细胞对铁需求量增加,ROS水平明显升高,正是这种对铁的高依赖性和高水平ROS,使得肿瘤细胞更容易发生铁死亡[1]。因此,诱导肿瘤细胞铁死亡成为一种新型的抗肺癌治疗策略。本文将对铁死亡调节机制、铁死亡与肺癌的关系作一综述,以期为肺癌的治疗提供理论基础。
1 铁死亡的调节机制
铁代谢、氨基酸和谷胱甘肽代谢以及脂质代谢是铁死亡的3大生化过程。
1.1 铁代谢
铁在食物中主要以Fe3+形式存在,经肠道铁还原酶如细胞色素B、血红素加氧酶1(HO-1)等还原成Fe2+,并在二价金属转运蛋白1(divalent metal transporter 1,DMT1)的作用下转运至小肠上皮细胞(intestinal epithelial cell,IEC),被IEC吸收[2]。在铁死亡过程中DMT1表达上调[2]。IEC所吸收的铁(Fe2+)在膜铁转运蛋白(ferroportin 1,FPN1)的作用下被运输至细胞外,并在肠细胞基地外侧被多铜氧化酶蛋白氧化为Fe3+,与转铁蛋白(transferrin,TF)结合形成TF-Fe3+复合物,经血液循环,运输至各组织与脏器[2]。循环中的TF-Fe3+与细胞膜表面上的转铁蛋白受体1 (transferrin receptor 1,TFR1)结合,经胞吞作用进入细胞,Fe3+被释放,继而被前列腺六跨膜表皮抗原3(six-transmembrane epithelial antigen of prostate 3,STEAP3)还原为Fe2+,经DMT1进入细胞质[2-3](图1)。细胞质中的Fe2+称为不稳定铁池,具有代谢活性,在多种生物功能中发挥作用,如凋亡、坏死、铁死亡等。当细胞内铁过载和抗氧化能力不足时,游离的Fe2+,一方面,通过芬顿反应直接催化脂质过氧化物,产生大量羟自由基,激起强烈的氧化应激反应,产生大量的ROS,诱发铁死亡[2];另一方面,作为辅助因子,增强各种代谢酶(如LOX家族脂氧合酶、PDH1)活性,促进脂质ROS的生成[2]。因此,铁是铁死亡的必要元素,铁代谢是铁死亡的必要过程。
1.2 氨基酸和谷胱甘肽代谢
谷氨酸/胱氨酸转运体system Xc-,作为跨膜蛋白,由两个亚基组成——轻链SLC7A11(也称为xCT)和重链SLC3A2(也称为CD98)[1](图1)。xCT为其主要功能亚基,由SLC7A11基因编码合成,对胱氨酸和谷氨酸有高度的特异性,负责主要的转运活动;SLC3A2,主要作为伴侣蛋白,维持xCT蛋白的稳定性。System Xc-调控着胞外胱氨酸和胞内谷氨酸以1∶1比例交换进出细胞[1]。谷氨酸(glutamic acid,Glu)、半胱氨酸(cysteine,Cys)和甘氨酸(glycine,Gly)在谷氨酸-半胱氨酸连接酶(glutamate cysteine ligase,GCL)和谷胱甘肽合成酶(glutamylcysteine synthetase,GSS)的催化下,生成还原型谷胱甘肽(glutathione,GSH)(图1)。谷胱甘肽过氧化物酶(glutathione peroxidases,GPXs)是一种进化上高度保守的酶,以GSH为辅助因子,将过氧化物(如R-OOH)还原为相应的醇(如R-OH),从而限制铁依赖的有毒自由基的形成(如R-O·),抑制脂质ROS的生成[1](图1)。GPX4是铁死亡中最核心的调控因子,胞内GSH含量直接影响GPX4酶活性。
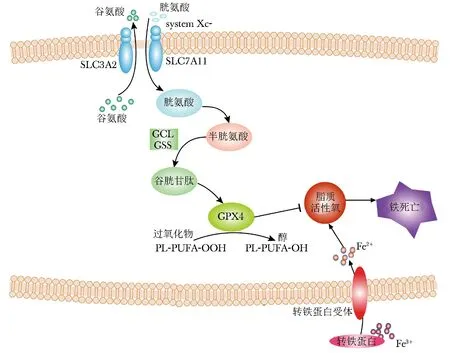
图1 铁死亡调节机制示意图 Fig 1 Regulatory mechanism of ferroptosis
1.3 脂质代谢
脂质过氧化是指自由基或非自由基等氧化剂从多不饱和脂肪酸(polyunsaturated fatty acids,PUFAs)的二烯丙基亚甲基群中获取一个不稳定的氢原子,通过氧化作用生成大量脂质过氧化自由基和过氧化氢的过程[4]。细胞内PUFAs的含量决定着细胞脂质过氧化程度以及对铁死亡的敏感性。细胞经铁死亡诱导剂erastin处理后,PUFAs花生四烯酸等和PUFA衍生物烟油酸盐等含量明显减少[5]。许多参与调控脂肪酸合成的因子和信号分子,如谷氨酰胺分解反应、柠檬酸合成酶和乙酰辅酶A羧化酶等脂氧合酶,通过介导脂质氧化参与调控铁死亡过程[6]。
酯酰基辅酶A合成酶长链家族成员4(acyl-CoA synthetase long-chain familymember4,ACSL4)是铁死亡脂质代谢的助力者。一方面,ACSL4和磷脂胆碱酰基转移酶3(lysophosphatidylcholine acyltrans-ferase 3,LPCAT3)将游离的长链多不饱和脂肪酸活化,促进溶血卵磷脂转换为卵磷脂,参与氧化细胞膜磷脂质的合成,进而介导铁死亡过程[7];另一方面,ACSL4将花生四烯酸辅酶A酯化成酰基辅酶 A(coenzyme A,CoA),用于脂肪酸氧化和铁死亡所需多不饱和脂肪酸的生物合成[7]。CoA的缺失使得脂质过氧化底物减少,铁死亡程度下降[7]。在PUFAs相关的磷脂质中,含有花生四烯酸(arachidonoyl,AA)或肾上腺酸(adrenoyl,AdA)的磷脂酰乙醇胺(phosphatidylethanolamines,PEs),是铁死亡中脂质氧化作用的关键底物,能够被15-脂氧合酶(15-LOX)氧化生成脂质过氧化氢(H2O2),促进铁死亡[8]。当ACSL4基因敲除或功能抑制时,AA或AdA酯化过程受阻,细胞内脂质过氧化物产生减少,铁死亡被抑制[7]。
2 铁死亡与肺癌的关系
2.1 铁死亡与肺癌发生发展
2.1.1 铁离子:流行病学和实验室研究证实,铁超载与肺癌的发生发展有关,高铁摄入量与肺癌风险之间存在显著正相关性。一项临床试验数据表明,肺癌患者的血清铁、铁蛋白、总铁结合力明显高于健康对照组,血清铁浓度越高,患肺癌风险越大[9]。与之结果一致的是,台湾一项研究对2018年至2009年309 443名的招募时,非肿瘤人群进行中位随访时间为7.07年的随访,其中8 060例确诊肿瘤,3 066例因肿瘤死亡,高血清铁(>120 μg/dL)增加了恶性肿瘤的发病与死亡风险,且与肿瘤发病率与病死率成正相关[10]。大量基础研究表明,过量的铁会诱发凋亡、坏死和铁死亡[3]。铁死亡诱导剂erastin促进ROS的累积和细胞死亡,外源性铁显著增强erastin所诱导的细胞死亡,而铁离子螯合剂(deferoxamine,DFO)能够逆转erastin所引起的细胞死亡现象[1]。在裸鼠肺癌肿瘤模型中,过表达转铁蛋白受体1(transferrin receptor 1,TFR1加快肺癌细胞对铁的吸收速度,促进肿瘤生长,缩短小鼠生存期[11]。热休克蛋白B1(heat shock protein B1,HSPB1)通过抑制TFR1循环,降低细胞内铁离子浓度;HSPB1的失活有助于铁的积累,促进erastin所诱导的肿瘤细胞铁死亡[12]。值得一提的是,虽然铁可以通过芬顿反应促进脂类ROS生成,但其他途径(如H2O2)造成的ROS累积并不会引起铁死亡。因此,铁在铁死亡中的作用机制和应用方面的许多问题仍然没有答案。
2.1.2 SLC7A11:SLC7A11为一种潜在的肺癌生物标志物,与癌旁组织相比,SLC7A11在NSCLC组织中高表达,与生存期成负相关[13]。在体内外,SLC7A11均能促进肺癌细胞的增值与转移,敲减SLC7A11可逆转上述现象[13]。在人肺腺癌细胞系A549中,SLC7A11通过介导胱氨酸摄取帮助肺癌细胞在细胞应激下重建氧化还原稳态,减少ROS的生成,具有促进肿瘤的作用;反之,siRNA干扰敲低SLC7A11表达,降低细胞内GSH含量,抑制A549细胞增殖[13]。
在KARS突变型肺腺癌患者中,SLC7A11高表达,与肺癌进展呈正相关[14]。与之对应的是,在KARS突变的肺腺癌细胞系中,胞内胱氨酸、GSH含量较高。敲除SLC7A11基因或阻断SLC7A11功能,能够降低胞内胱氨酸摄取、抑制细胞内GSH的生物合成,在体外显著抑制肿瘤生长与转移、延长小鼠生存期,在体内选择性杀伤KARS突变的肺癌细胞[14]。KARS突变型肺腺癌细胞对SLC7A11的缺失更为敏感,这为KARS突变肺癌的治疗带来希望。
2.1.3 GPX4:GPX4在癌组织中的表达高于正常组织,与肺癌TNM分期、淋巴转移和远处转移成正相关,与患者预后、生存期呈负相关;肺癌细胞系亦呈现GPX4高表达状态[15]。过表达GPX4能够促进肺癌细胞增殖,抵抗铁死亡;反之,siRNA 敲减GPX4表达或RSL3抑制GPX4活性,抑制H1299、A549和NCI-H460细胞增殖、迁移、侵袭,而铁死亡抑制剂ferrostatin-1(Fer-1)可逆转上述现象[15]。这意味着抑制GPX4能够诱导肺癌细胞发生铁死亡,靶向GPX4可能是一种新的肺癌治疗模式。
2.1.4 FSP1:FSP1是一种独立于经典GPX4信号通路的铁死亡抑制因子和非线粒体CoQ抗氧化剂系统的关键成分[16]。当肺癌细胞GPX4基因缺失时,FSP1被豆蔻酰化修饰,利用NAD(P)H还原CoQ10,生成亲脂性自由基捕获抗氧化剂(radical-trapping antioxidants,RTA)阻止脂质过氧化,从而抑制铁死亡[16]。FSP1表达水平越高,肺癌细胞铁死亡抵抗程度越大,而FSP1抑制剂(iFSP1)可逆转FSP1所致的铁死亡抵抗,增加肺癌细胞对铁死亡的敏感性,促进肺癌细胞发生铁死亡[16]。目前,对于FSP1的研究还处于萌芽阶段,后续还需进一步研究。
2.2 铁死亡与化疗药物耐药
顺铂(cisplatin,DDP)通过促进脂质过氧化,升高MDA、ROS,促进HO-1和NQO-1的表达,诱导肺癌细胞铁死亡,而这一过程可被Fer-1所抑制[17]。Nrf2/xCT通路的激活是NSCLC细胞耐顺铂的主要机制之一。Erastin和索拉菲尼通过抑制Nrf2下游靶基因xCT的表达,耗竭GSH,诱发铁死亡,降低细胞活性,增强NSCLC细胞对顺铂的敏感性[18]。相反,过表达SLC7A11增强肺癌细胞对顺铂的耐药性[18]。将SLC7A11的表达与1 400种候选抗癌药物的效力联系起来,其中,与39种药物药效呈正相关,与296种药物药效呈负相关,提示SLC7A11可作为谷胱甘肽介导的抗癌药物耐药性的预测因子,预测多种化学药物敏感性[19]。
与A549细胞相比,A549-DDP细胞(A549顺铂耐药株)高表达GPX4[20]。抑制GPX4可增强顺铂的细胞毒性作用;反之,过表达GPX4导致顺铂细胞毒性减弱[20]。与单纯顺铂或GPX4特异性抑制剂RSL3治疗相比,顺铂联合RSL3显著抑制了H1299和A549细胞活性、迁移与侵袭,MDA、ROS、脂质过氧化物含量升高,提示RSL3可增强顺铂的敏感性[20-21]。
此外,铁自噬被证实促进癌细胞铁死亡。在这一过程中,铁蛋白降解,铁离子从内涵体释放到细胞质内不稳定的铁池中,从动态铁池释放的过量的铁通过芬顿反应,产生大量的ROS,诱发铁死亡。顺铂处理肺癌细胞所引起的细胞内铁离子浓度、MDA和ROS含量升高、铁蛋白(ferritin 1,FTH1)表达下降,被自噬抑制剂3-MA所逆转,提示顺铂能够诱发铁自噬[20]。在体外,与顺铂组相比,顺铂联合RSL3组FTH1水平下降,自噬标志物LC3B Ⅱ/LC3BⅠ比值升高、P62蛋白水平下降,细胞内铁离子浓度和MDA含量增加[20]。总之,上述现象表明,顺铂能够通过介导铁自噬,诱发铁死亡。
2.3 铁死亡与放疗抵抗
经放射治疗(ionizing radiation,IR;简称放疗)处理后,NSCLC细胞ROS含量升高,ACSL4、SLC7A11、GPX4表达升高,线粒体缩小,膜密度增强,为典型的铁死亡形态学特征;铁死亡抑制剂Fer-1可逆转IR所引起的细胞死亡,提高NSCLC细胞活性[22]。采用CRISPR/Cas9技术沉默H460和A549细胞中ACSL4表达后,ACSL4的缺失显著减弱了erastin所诱导的肺癌细胞铁死亡,促进放疗抵抗[22]。过表达SLC7A11或GPX4基因削弱IR所诱导的脂质过氧化反应,降低铁死亡标志基因PTGS2的表达,抑制铁死亡,增强NSCLC细胞的放疗抵抗性[22]。与正常NSCLC细胞相比较,GPX4在放疗抵抗性NSCLC细胞中表达明显升高[23]。RNA干扰技术沉默GPX4后,放疗抵抗性A549(A549-R)和H460(H460-R)对铁死亡的敏感性增强[23]。因此,铁死亡激活剂erastin能够增强A549-R和H460-R细胞对放疗的敏感性,降低NSCLC细胞对放疗的耐药性,促进细胞死亡;反之,铁死亡抑制剂DFO可部分“挽救”erastin所诱导的细胞死亡[23]。MicroRNA(miRNA)是一种非编码RNA,参与调控多种癌基因表达,在放疗抵抗性NSCLC细胞中,miR-7-5p表达升高,miR-7-5p通过下调线粒体铁转运蛋白,降低Fe2+浓度,减弱芬顿反应,降低细胞内ROS含量,抑制铁死亡,增强细胞放射抵抗性[24]。
2.4 铁死亡与免疫治疗
T细胞介导的细胞免疫在肿瘤发生发展中过程中发挥重要作用。在免疫治疗过程中活化的CD8+T细胞能够增强肿瘤细胞内铁死亡特异性的脂质过氧化反应;反之,铁死亡的激活有助于免疫治疗的抗肿瘤效果[25]。CD8+T细胞释放的IFN-γ下的表达,抑制胱氨酸的摄取,促进脂质过氧化和铁死亡[25]。耗竭胞内胱氨酸或阻断PD-L1免疫检查点,显著增强T细胞介导的抗肿瘤免疫,诱导肿瘤细胞铁死亡[25]。同时,临床数据显示,在黑色素患者中,胱氨酸相关转运蛋白SLC7A11和SLC3A2的表达与CD8+T细胞的数量、IFN-γ的表达水平以及患者的预后成负相关[25]。虽然目前针对肺癌,T细胞与铁死亡的关系未明确指出,但是,不难发现T细胞促进肿瘤细胞铁死亡是一种潜在的治疗方法,有助于增强免疫治疗疗效。
3 总结
铁死亡,作为一种新发现的细胞死亡形式,在肿瘤治疗中表现出独特的优势和巨大的潜力。许多侵袭性和抗药性的癌细胞对铁死亡的敏感性,以及美国FDA批准六甲蜜胺(altretamine)、索拉菲尼(sorafenib)、二氧化硅纳米颗粒(silica nanoparticles)作为铁死亡诱导剂用于肿瘤治疗,这使得人们对铁死亡的治疗潜力产生了很高的期望。虽然,近年来铁死亡相关研究取得了巨大的进展,但仍有一些悬而未决的问题有待解决,如铁死亡中ROS的特殊性、铁死亡在免疫治疗中的具体作用等。此外,不同组织间细胞对铁死亡的敏感性存在很大的差异,对铁死亡诱导剂索拉菲尼、erastin等敏感性也具有显著的个体间差异。因此,寻找能够反映细胞、个体对铁死亡敏感性的生物指标、发现新的铁死亡诱导剂,对于提高对铁死亡相关疾病的认识、肺癌诊疗水平具有重要意义。铁死亡将成为肿瘤治疗的一种新策略,打破目前肺癌治疗的瓶颈,为肺癌患者带来利益。
