高效液相色谱法测定乳粉中三聚氰胺的方法优化
2021-03-22卢竹阳沈蕾陈丹莹柏宁
卢竹阳 沈蕾 陈丹莹 柏宁
三聚氰胺含氮量高,俗称“伪蛋白”,几乎无味,微溶于水,可溶于甲醇、乙酸、甲醛等,经常被用作化工原料,对健康有害,我国明确规定三聚氰胺不是食品原料,也不是食品添加剂,禁止人为添加。“三鹿奶粉”事件影响恶劣,也给食品检验机构敲响警钟,三聚氰胺参数在乳品检测中成了必检指标,研究其检测方法尤为重要。
GB/T 22388-2008《原料乳与乳制品中三聚氰胺检测方法》明确规定,测定三聚氰胺有三种方法,即液相色谱-质谱/质谱法、气相色谱-质谱联用法和高效液相色谱法。前两种方法涉及的仪器高端精密,费用昂贵,后续维护保养较为繁琐,不具备普遍性,本文对高效液相色谱法检测乳粉中三聚氰胺的前处理和色谱条件进行了优化。
一、材料和方法
1.仪器。Agilent-1200高效液相色谱仪(VWD),多功能氮吹仪,高速冷冻离心机,数控超声波清洗器,电子天平(Ⅰ级),固相萃取联用仪,SPE小柱:Poly-Sery MCX,60mg/3mL。
2.试剂。三聚氰胺标样(1.0mg/mL),甲醇、乙腈、正己烷均为色谱纯,柠檬酸、辛烷磺酸钠、氨水、三氯乙酸均为分析纯。
3.样品前处理。(1)提取。准确称取1g乳粉置于50ml具塞离心管中,加入8ml三氯乙酸溶液(1%)和2ml乙腈,超声提取约10min,涡旋提取约10min,9500r/min离心8min,取上清液于50ml离心管中;残渣重复提取一次,合并两次提取液,加入10ml三氯乙酸溶液和饱和的正己烷溶液,涡旋振荡1min,9500r/min离心6min,取下层清液,待凈化。(2)净化。依次用3ml甲醇和5ml水对SPE小柱进行活化和平衡,抽至近干;将待净化的清液转至SPE小柱中,用3ml水和3ml甲醇进行淋洗,抽至近干;用6mL氨化甲醇溶液(5%)洗脱,洗脱液于50℃氮气吹干,残留物用1ml流动相定容,涡旋混合1min,过0.22μm微孔水相滤膜后,供测定。
4.色谱参考条件。色谱柱:Agilent Eclipse XDB-C18柱,4.6mm×250mm,5μm。流动相:离子对试剂缓冲液(称取2.16g辛烷磺酸钠和2.10g柠檬酸于烧杯中,加入约980ml水溶解,调节pH3.0,转移至1L容量瓶,定容)-乙腈(93+7),混匀。流速:1.0ml/min,柱温:40℃,波长:240nm,进样量:20μL。
5 .标准曲线的绘制。用流动相将标准储备液(1.0mg/mL)逐级稀释成0.5、2.0、10.0、20.0和40.0mg/kg标准工作液,低浓度到高浓度依次进样。以峰面积为纵坐标、浓度为横坐标作曲线,得到标准曲线回归方程,即y=84.47216x+6.65842,r2=0.99998,符合检测要求。
二、结果与分析
1.前处理方法优化。在乳粉前处理过程中,将国标中的振荡提取优化成涡旋提取,让蛋白质等大分子物质沉淀更充分。这样做减少了过滤的环节,减少了目标物损失,并增加了二次提取步骤,将目标物充分提取,增加回收率。分别在空白乳粉中添加标样(10.0mg/kg)1ml和2ml,检测到的三聚氰胺含量分别为9.23mg/kg和13.24mg/kg,回收率分别为92.3%和93.2%,满足检测要求。
2.色谱条件优化。根据GB/T 22388-2008国标要求,流动相:离子对试剂缓冲液-乙腈(90+10),混匀,按此条件进样,三聚氰胺于9.27min出峰,查看光谱图,会发现此峰前半部分和后部分是两个不同的光谱,详见图1、图2。为了分开两物质,将流动相比例优化为93+7,混匀,按此条件进样,进样时间调至16min,查看光谱后确认两物质出峰时间为11.833min和14.034min,成功分离,所以93+7的液相比例更能准确测得三聚氰胺含量。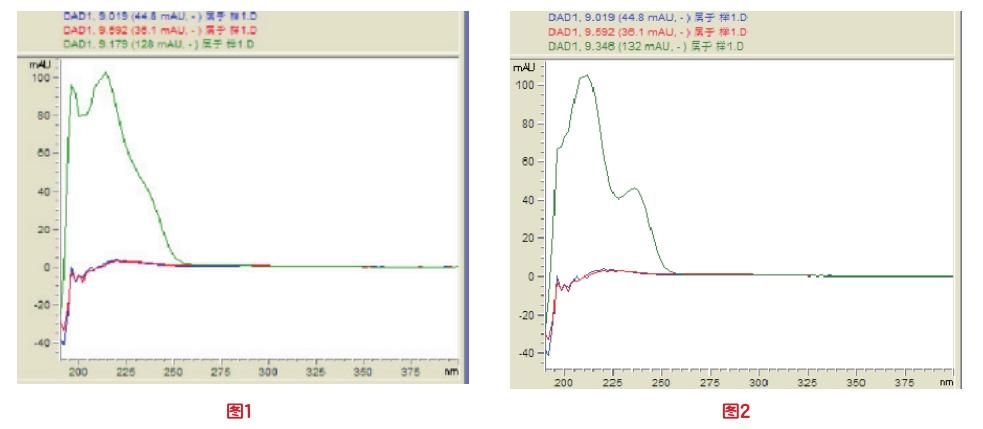
本文在国家标准GB/T 22388-2008的基础上,对高效液相色谱法的前处理过程和色谱条件进行了优化,优化后的回收率高达93%,并成功解决了两物质重叠峰的问题,标准工作曲线、质控样检测和加标回收等一系列的质量控制实验均表明优化后的方法具有可行性,对于检测乳粉中三聚氰胺残留量具有实际的应用价值。
