一种检测纽莫康定B0和C0质量浓度的HPLC方法研究
2021-03-18魏荣省李旭娇李会会李心怡韩守暖朱兵峰
魏荣省,李旭娇,李会会,李心怡,韩守暖,朱兵峰
(1.鲁南制药集团股份有限公司,山东 临沂 273400;2.鲁南新时代生物技术有限公司,山东 临沂 273400;3.哺乳动物细胞高效表达国家工程实验室,山东 临沂 273400)
纽莫康定(Pneumocandins)是由Zalerionarboricola[1-5]产生的一类天然抗真菌药物,对多种念珠菌、地方性真菌、曲霉及卡氏肺囊虫均有效,受到人们的广泛关注。其主要作用机理是能够非竞争性的抑制真菌细胞壁中β-1,3-葡聚糖合成酶的活性,引起细胞壁裂解以及细胞内外渗透压的改变,从而将真菌细胞杀死。按照其结构中脯氨酸上取代基的不同主要分为纽莫康定A0(3-羟基-4-甲基脯氨酸)、纽莫康定B0(3-羟基脯氨酸)和纽莫康定C0(4-羟基脯氨酸)3大类[6],其中纽莫康定B0脱侧链后的环状六肽母核是合成棘白菌素类药物醋酸卡泊芬净[7](Caspofungin acetate)的中间体。醋酸卡泊芬净由默克公司原研,属广谱抗真菌药物,对包括曲霉和念珠菌属在内的真菌均有良好的抗菌作用。纽莫康定B0与纽莫康定C0属于同分异构体,两者的差别仅在于脯氨酸残基上的一个羟基的位置不同,在通常条件下很难实现分离,而纽莫康定C0为纽莫康定B0生产过程中的关键杂质,会通过合成路线逐步传递到终产品醋酸卡泊芬净原料药中,并生成非药效成分类卡泊芬净,影响产品质量。目前,各国药典虽然还未收录纽莫康定和醋酸卡泊芬净相关质量标准,但是各生产厂家均已将纽莫康定C0列入质量标准加以控制。因此,开发一种能同时准确检测纽莫康定B0和纽莫康定C0质量浓度的方法对于控制纽莫康定乃至醋酸卡泊芬净的产品质量非常重要。
反相HPLC法[8-10]测定纽莫康定B0的文献报道虽然较多,但此类方法只能实现纽莫康定A0的分离,纽莫康定B0和纽莫康定C0峰完全重叠;通过正相HPLC法,虽然能够实现纽莫康定B0和纽莫康定C0的同时检测,但使用的二氯甲烷、乙酸乙酯等试剂毒性较大,杂质检出少且峰响应值偏低。亲水作用色谱法HILIC模式[11]为新兴的色谱技术,使用两性离子填料[12]能够同时检测纽莫康定B0和纽莫康定C0,但此填料柱效差,键合相流失快,色谱柱寿命短,且要求使用等度洗脱,残留杂质可能会影响下一样品的检测;使用氨基填料不能分离纽莫康定B0和纽莫康定C0;使用聚合多氨填料能够同时检测纽莫康定B0和纽莫康定C0,但柱寿命只比氨基柱延长50%左右,同样寿命短。笔者使用三键键合酰胺填料,建立了HILIC模式的梯度方法,既能同时快速准确测定纽莫康定B0和纽莫康定C0,又大大延长柱寿命,也解决了样品批处理的残留问题,为有效控制纽莫康定生产质量提供了一条有效途径。
1 仪器与材料
1.1 仪 器
Agilent 1260 Infinity Ⅱ高效液相色谱仪(VWD-DAD)、Agilent 1290 2DLC-6540B QTOF LC-MS、Thermo Evolution 3000紫外分光光度计、METTLER TOLEDO XS105DU分析天平、METTLER TOLEDO MS 205 DU分析天平。
1.2 材 料
纽莫康定B0对照品(质量分数95.56%,批号16001,大同市矿区风华生物科技有限公司),纽莫康定C0对照品(质量分数95.11%,批号16002,大同市矿区风华生物科技有限公司);纽莫康定B0发酵液(F 301180301 F,自制),纽莫康定B0粗品(F 301170401 Ⅰ,自制),纽莫康定B0冻干粉(F 301180501 K,自制);乙腈、甲醇和乙醇均为色谱纯,盐酸、双氧水和氢氧化钠均为分析纯,水为纯化水。
2 方法与结果
发酵过程中会产生一系列纽莫康定类似物,其中3个主要纽莫康定结构类似物结构式为

本方法主要是针对其中同分异构体纽莫康定B0和C0进行的研究。
2.1 色谱条件
色谱柱Waters XBridge®Amide柱(4.6 mm×250 mm,3.5 μm),检测波长220 nm,流速1 mL/min,柱温40 ℃,进样量10 μL;流动相为V(A)∶V(B)(其中:A为纯水;B为乙腈),梯度洗脱(0~10 min,B 93%~84%;10~18 min,B 84%;18~19 min,B 84%~50%;19~28 min,B 50%;28~29 min,B 50%~93%;29~35 min,B 93%)。
2.2 溶液配制
2.2.1 纽莫康定B0对照品溶液
取纽莫康定B0对照品约20 mg,精密称定,置于50 mL容量瓶中,加乙醇超声溶解后,稀释至刻度,摇匀,制得质量浓度为400 mg/L的纽莫康定B0对照品溶液。
2.2.2 纽莫康定C0对照品溶液
取纽莫康定C0对照品约20 mg,精密称定,置于50 mL容量瓶中,加乙醇超声溶解后,稀释至刻度,摇匀;精密量取上述溶液1 mL,置于10 mL容量瓶中,用乙醇稀释至刻度,摇匀,制得质量浓度为40 mg/L的纽莫康定C0对照品溶液。
2.2.3 纽莫康定B0发酵液
准确量取纽莫康定发酵液20 mL,加乙醇80 mL,匀浆10 min,过滤,取滤液适量离心(4 000 r/min)10 min,取上清液,用0.45 μm滤膜过滤,即得。
2.2.4 纽莫康定B0粗品溶液
取粗品约20 mg,精密称定,置于50 mL容量瓶中,加乙醇超声溶解后,稀释至刻度,摇匀,即得。
2.2.5 纽莫康定B0冻干粉溶液
取纽莫康定B0冻干粉约20 mg,精密称定,置于50 mL容量瓶中,用乙醇超声后,稀释至刻度,摇匀,即得。
2.3 色谱条件的选择
2.3.1 波长的确定
实验分别采用紫外分光光度计和二极管阵列检测器对纽莫康定B0对照品溶液、纽莫康定C0对照品溶液分别进行UV全波长扫描,扫描纽莫康定B0乙醇溶液UV光谱图如图1所示。结果显示:在乙醇溶液中纽莫康定B0的UV吸收和纽莫康定C0的UV吸收光谱图完全一致,在波长220 nm处可以看到有较强吸收,且响应相对平缓,波长耐用性较好。因此,笔者选用220 nm作为纽莫康定B0和纽莫康定C0质量浓度的检测波长。

图1 纽莫康定B0乙醇溶液UV光谱图
2.3.2 梯度洗脱优化
与甲醇水体系相比,乙腈水体系各杂质检出个数和峰形均较好,故选用乙腈水体系进行优化。首先通过等度洗脱方法摸索,V(纯水)∶V(乙腈)=8∶92为纽莫康定B0、纽莫康定C0洗脱的临界比例。为增加样品洗脱效果,保证纽莫康定B0、纽莫康定C0与前后杂的基线分离,选择V(纯水)∶V(乙腈)=7∶93为梯度洗脱初始比例,V(纯水)∶V(乙腈)=16∶84为主峰洗脱比例。由于样品中残留杂质会影响下一针样品的准确度,经不同梯度摸索,得出V(纯水)∶V(乙腈)=50∶50能够对样品的杂质进行完全洗脱,所以选用V(纯水)∶V(乙腈)=50∶50进行杂质洗脱,为保证批处理样品时,连续进样系统的稳定性,增加V(纯水)∶V(乙腈)=7∶93系统平衡,用时6 min。
2.4 LC-MS鉴定
取纽莫康定B0粗品溶液,按照2.1项中色谱条件采用分流法进行LC-MS分析,其结果如图2所示,纽莫康定B0和纽莫康定C0保留时间分别为9.04 min和10.16 min,m/z均为1 064[13-14]。
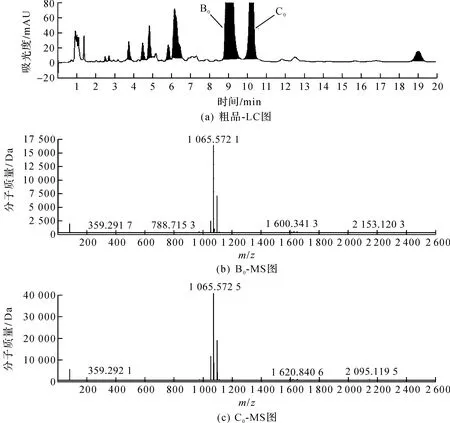
图2 LC-MS图
2.5 分析方法验证
笔者对本方法耐用性、系统适用性、专属性、线性、检测限、定量限、准确度、重复性和溶液稳定性进行了相应验证,具体验证结果汇总如表1所示。
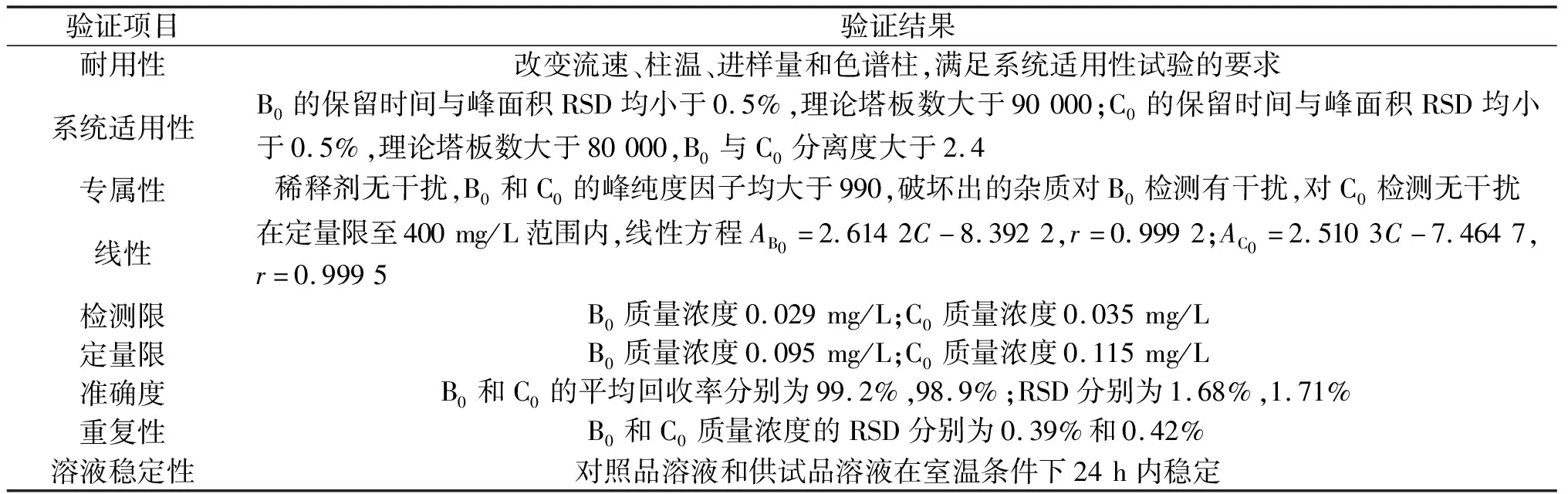
表1 方法验证结果汇总
2.5.1 耐用性试验
通过改变流速(0.8,1.2 mL/min)、柱温(35,45 ℃)、进样量(5,15 μL)、色谱柱(Waters XBridge®Amide柱其他批次和月旭Ultimate®HILIC Amide柱),考察方法耐用性。经试验,以上色谱条件变化均能满足最新版中国药典对系统适用性试验的要求,提示该方法耐用性良好。
2.5.2 系统适用性试验
取粗品溶液,按照2.1项中色谱条件进行测定,连续进样6针,记录色谱图如图3所示。结果显示:纽莫康定B0色谱峰的保留时间与峰面积的RSD均小于0.5%,理论塔板数大于90 000;纽莫康定C0色谱峰的保留时间与峰面积的RSD均小于0.5%,理论塔板数大于80 000,纽莫康定B0与纽莫康定C0色谱峰分离度大于2.4。
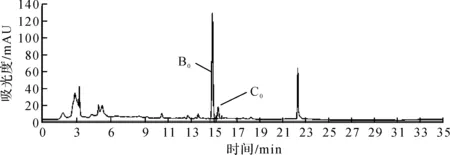
图3 典型色谱图
2.5.3 专属性试验
稀释剂色谱乙醇对纽莫康定B0和纽莫康定C0检测无干扰,溶解性良好,进样量30 μL以内无溶剂效应。液相DAD检测器显示纽莫康定B0和纽莫康定C0峰纯度因子均大于990。破坏试验测定结果如表2所示。

表2 破坏试验结果
破坏出的杂质对纽莫康定B0检测有干扰,对纽莫康定C0检测无干扰,主要降解杂质经LC-MS鉴定,m/z为1 064。
对品种最大单杂B0丝氨酸同系物进行定位,发现B0丝氨酸同系物与纽莫康定B0保留时间一致。
2.5.4 线性试验
分别精密量取纽莫康定B0和纽莫康定C0对照品溶液1,2,4,6,8,10 mL,置于6个10 mL容量瓶中,加乙醇稀释至刻度,定容,摇匀,制成6个不同质量浓度的线性溶液[15-16],加定量限溶液共7个点,按照2.1项中色谱条件,分别进样,记录色谱图。以对照品溶液中各组分质量浓度C为横坐标,色谱峰面积A为纵坐标,进行纽莫康定B0和纽莫康定C0的线性回归,得线性方程分别为
AB0=2.614 2C-8.392 2,r=0.999 2
AC0=2.510 3C-7.464 7,r=0.999 5
2.5.5 定量限、检测限试验
分别将纽莫康定B0和纽莫康定C0对照品溶液逐步稀释,按照2.1项中色谱条件进样测定,以信噪比约等于3的质量浓度为各组分的检测限,信噪比约等于10的质量浓度为各组分的定量限。测定结果如表3所示。

表3 定量限和检测限
2.5.6 准确度试验
各取纽莫康定B0对照品溶液和发酵液上清液0.5 mL混合(相当于100%水平的质量浓度400 mg/L)6份样品作为纽莫康定B0加标回收率供试品溶液[17],另各取纽莫康定C0对照品溶液和发酵液上清液0.5 mL混合(相当于100%水平的质量浓度40 mg/L)6份样品作为纽莫康定C0加标回收率供试品溶液,按照2.1项中色谱条件分别进样测定,按外标法以峰面积计算纽莫康定B0和纽莫康定C0的回收率。结果可得:纽莫康定B0和纽莫康定C0的平均回收率分别为99.2%,98.9%;相对标准偏差RSD分别为1.68%,1.71%。
2.5.7 重复性试验
精密称定粗品6份,按照2.2项中溶液配制方法配制供试品溶液,按照2.1项中色谱条件进样测定,按外标法以峰面积计算6份供试品中纽莫康定B0和纽莫康定C0质量浓度,计算纽莫康定B0和纽莫康定C0质量浓度的相对标准偏差RSD分别为0.39%,0.42%。
2.5.8 溶液稳定性试验
取纽莫康定B0对照品溶液、纽莫康定C0对照品溶液、发酵液、粗品溶液和纽莫康定B0冻干粉溶液,室温放置,于0,2,4,8,12,24 h分别进样测定,计算得纽莫康定B0对照品溶液主峰面积的RSD为0.75%,纽莫康定C0对照品溶液主峰面积的RSD为0.81%,发酵液中纽莫康定B0和纽莫康定C0峰面积的RSD分别为1.39%和1.44%,粗品溶液中纽莫康定B0和纽莫康定C0峰面积的RSD分别为1.27%和1.36%,纽莫康定B0冻干粉溶液峰面积的RSD为0.77%。结果表明对照品溶液和供试品溶液在室温条件下24 h内稳定。
3 结 论
杂质研究及控制是药品质量保证的关键因素,而异构体的控制是产品质量控制的难点。本实验同时检测纽莫康定样品中同分异构体纽莫康定B0和纽莫康定C0质量浓度的HPLC方法,并分别检测了纽莫康定发酵液、粗品和冻干粉。结果表明:样品中纽莫康定B0和纽莫康定C0的分离度、重复性和稳定性良好。此方法具有操作简单、稳定性强、灵敏度高和耐用性好等特点,为有效控制纽莫康定产品质量提供了保障。
