Ru基催化剂载体表面改性对草酸二甲酯加氢反应的影响
2021-03-12贺燕施建哲马奎岳海荣梁斌
贺燕,施建哲,马奎,岳海荣,梁斌
(四川大学 化学工程学院,四川 成都 610065)
以煤基合成气为原料,经草酸二甲酯(DMO)加氢制备高附加值产品乙醇酸甲酯(MG)的工艺路线具有重要意义[1-2]。目前针对DMO加氢催化剂的研究主要集中在钌基均相催化剂和铜基催化剂。钌基均相催化剂反应条件温和,MG收率高[3],但难以进行工业放大。而铜基加氢催化剂的工作温度较高(180~260 ℃)[4-6],MG易发生深度加氢[7-9]。此外,钌基多相催化剂也被广泛应用于温和的加氢反应体系[10-13]。活性炭(AC)具有稳定性好、比表面积大、导电性强等特点[14-17],常被用作电子型催化剂载体。
本文以AC为载体负载Ru,并采用氮掺杂、弱氧化和强氧化的方法对AC进行改性。系统研究了改性前后钌基催化剂的物化性质,并揭示了其对DMO加氢性能的影响。
1 实验部分
1.1 材料与仪器
活性炭(XFP-01);三水合氯化钌(Ru≥38%)、草酸二甲酯、甲醇、三聚氰胺、双氧水(40%)、硝酸均为分析纯;H2(99.99%);N2(99.99%)。
YZPR-100M微型反应釜;SP-2100A 气相色谱仪;GSL-1500X-50 管式炉;Spectrum Two 傅里叶变换红外光谱仪;LabRAM HR 拉曼激光光谱仪;VarioELcube 元素分析仪;PANalytical Empyrean X射线衍射仪;Tecnai-G20 透射电子显微镜;Thermo Escalab 250Xi X射线光电子能谱仪;DM-501 接触角测试仪。
1.2 催化剂的制备
氮掺杂选择以三聚氰胺为氮源,称取等比例的AC和三聚氰胺,研磨至混合均匀。在管式炉氮气氛围中800 ℃煅烧2 h,得到的样品标记为AC-N。
AC弱氧化改性选用双氧水。将1 g AC加入至30 mL浓度为40%的双氧水溶液中搅拌14 h。抽滤,用去离子水洗涤干净。在80 ℃烘箱中过夜烘干,制得的样品标记为AC-H2O2。
AC强氧化改性选用HNO3。将1 g AC加入至30 mL浓度为0.5 mol/L的硝酸溶液,在80 ℃下搅拌14 h。抽滤,用去离子水洗涤至滤液呈中性。在80 ℃烘箱中过夜烘干后,获得的样品标记为AC-HNO3。
Ru基催化剂采用浸渍法制备。称取一定量(Ru理论负载量为2%)的RuCl3·3H2O溶于水后,加入载体充分搅拌,在50 ℃下旋转蒸发除去水。在250 ℃下氢气氛围中进行预处理,即得到还原后的Ru/AC-X样品(X代表不同的载体改性方式)。
1.3 催化剂活性评价
催化剂的活性评价在100 mL小型高压反应釜中施行。将3.5 mmol草酸二甲酯溶于30 mL甲醇中,搅拌至完全溶解后,将混合液体和0.5 g催化剂一同装入反应釜中。密封好反应釜后充入氢气,置换反应釜中的空气。置换10次后充入氢气至反应压力5 MPa,升温至90 ℃反应24 h。反应完毕,离心分离催化剂。加氢产物通过气相色谱仪进行检测。
2 结果与讨论
2.1 活性炭载体表面结构
2.1.1 FTIR 采用FTIR对活性炭载体表面结构进行表征,结果见图1。
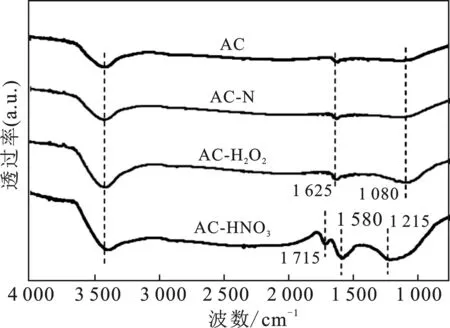
图1 AC及改性后AC-X样品的红外谱图Fig.1 FTIR spectra of the AC and AC-X samples

2.1.2 拉曼光谱 对于活性炭这种红外光谱敏感性不强的材料,常用拉曼(Raman)光谱来检测其表面官能团。在碳材料中,1 360 cm-1左右的峰为D带,其通常由缺陷处的sp3杂化的C产生。1 580 cm-1左右的峰为G带,通常由sp2杂化的C原子的面内伸缩振动产生。因此常用D带和G带的比值来描述碳材料的石墨化程度。碳材料的石墨化程度和导电性相关,越高的石墨化程度则有着更好的导电性。良好的导电性有利于电子在载体和金属活性中心之间的传递,从而有利于催化活性的提升。见图2,所有样品的ID/IG值均<1,G带的强度>D带,说明样品的石墨化程度较好。另外,改性后样品的石墨化程度均有所降低,意味着改性对样品的石墨化结构有一定的破坏。氮掺杂改性对样品的石墨化结构破坏程度最大,这是由于杂原子N的引入,形成了更多的缺陷位。而氧化改性由于在AC表面产生了更多的羟基和羧基,对其石墨化结构也有一定程度的破坏,但破坏程度较小。其中,强氧化改性对石墨化结构的破坏最小。
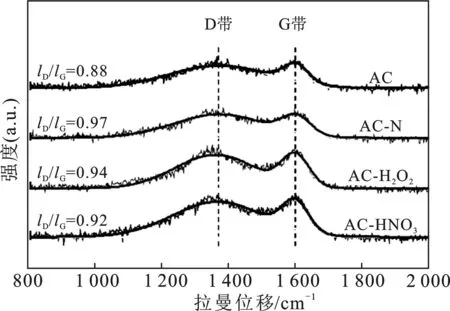
图2 AC及改性后AC-X样品的拉曼谱图Fig.2 Raman spectra of the AC and AC-X samples
2.2 载体化学组成
对载体进行元素分析,结果见表1。

表1 AC及改性后的AC-X样品中元素含量Table 1 Element contents of AC and AC-X
2.3 催化剂氧化还原能力
采用氢气程序升温还原(H2-TPR)对样品的还原能力进行测试,结果见图3。
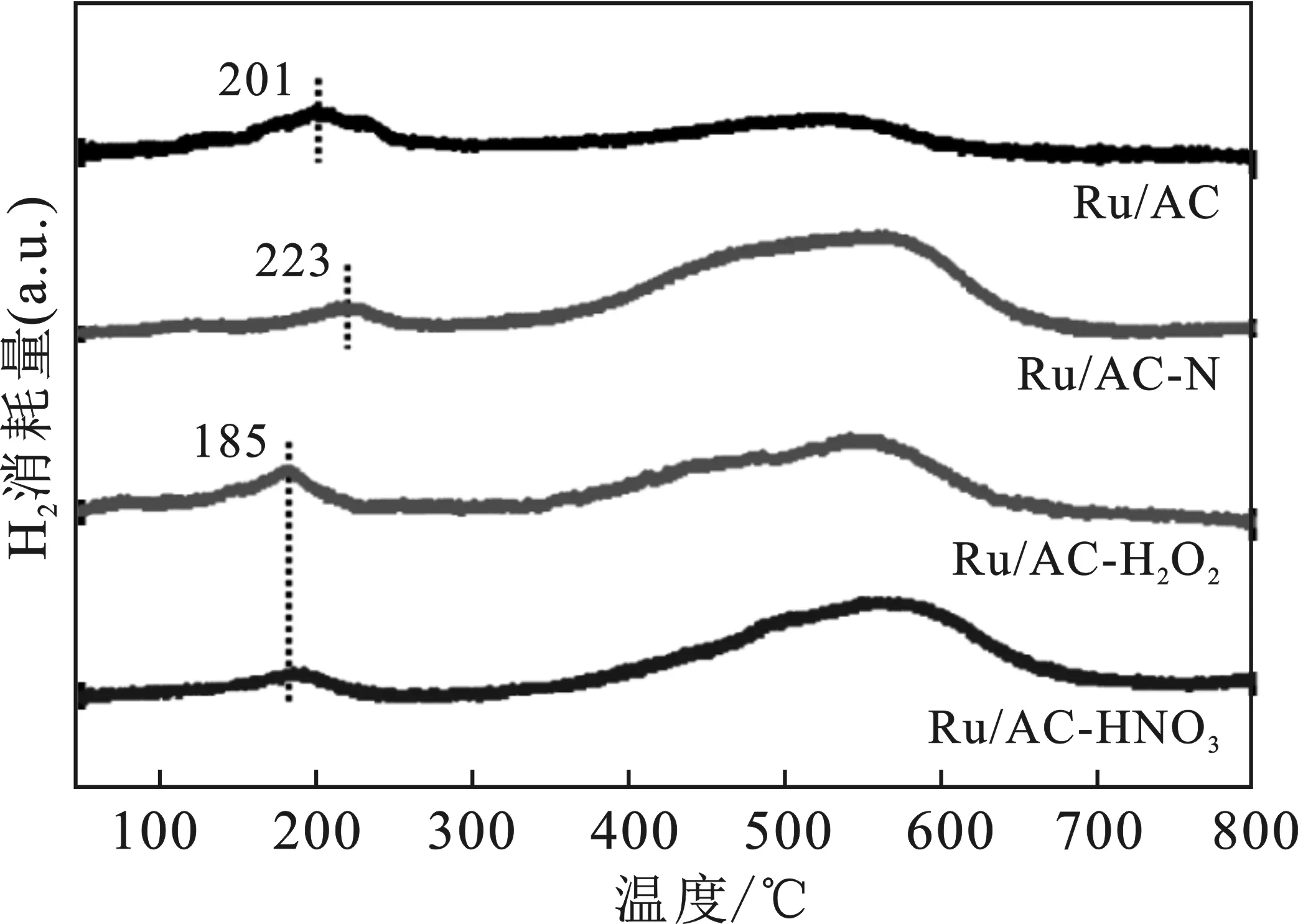
图3 Ru/AC、Ru/AC-N、Ru/AC-H2O2和Ru/AC-HNO3的H2-TPR谱图Fig.3 H2-TPR profiles of Ru/AC,Ru/AC-N,Ru/AC-H2O2,and Ru/AC-HNO3
由图3可知,Ru的还原峰位置一般位于200 ℃左右。Ru/AC的还原温度为201 ℃,氮掺杂改性之后还原温度升高到了223 ℃,说明载体和Ru之间的相互作用有所增强,不利于Ru的还原。氧化改性后,还原温度降低至185 ℃,可能是因为Ru元素在载体表面分散较好,因此可以在更低的温度下被还原为单质Ru。
此外,所有样品均在550 ℃左右出现了一个宽泛的峰,这是由于C在此温度下能和氢气发生反应,生成甲烷。同时,被还原成金属态的Ru也是良好的加氢催化剂,可以促进该反应的进行。Ru/AC在此处的峰相对较小,表示其加氢能力较差。为保证Ru完全还原为Ru0,同时避免载体上的碳和氢气反应生成甲烷,选择250 ℃作为催化剂的预还原温度。
2.4 催化剂活性金属晶体结构
采用X射线衍射对载体及负载Ru后催化剂的晶体结构进行表征,结果见图4。
图4 (a)AC及改性后AC-X样品的XRD谱图;(b)Ru/AC及Ru/AC-X催化剂的XRD谱图Fig.4 (a) XRD patterns of the AC and AC-X samples; (b) XRD patterns of the Ru/AC and Ru/AC-X catalysts
由图4可知,所有样品在24°和44°左右均出现馒头峰,可归属为无定形C的散射峰。在负载Ru前后,样品的XRD谱图几乎没有变化,说明Ru在不同方法改性的载体表面分散都很均匀,没有出现明显团聚,所有样品上Ru纳米颗粒的粒径均很小,低于了XRD仪器的检测限(<5 nm)。
2.5 催化剂微观形貌
采用高倍透射电子显微镜对催化剂微观形貌进行表征,结果见图5。

图5 催化剂的TEM,HRTEM以及粒径分布图Fig.5 TEM,HRTEM and the particle size distribution of the catalysts(a)(e)Ru/AC;(b)(f)Ru/AC-N;(c)(g)Ru/AC-H2O2;(d)(h)Ru/AC-HNO3
由图5可知,金属Ru以1~2 nm小颗粒的形式在载体表面均匀分散。采用统计学方法对载体表面的颗粒粒径尺寸进行分析。发现氮掺杂改性之后,Ru的平均粒径略有减小,从2.06 nm减小到1.91 nm。 这可能是由于Ru和载体之间存在相互作用,抑制了Ru在还原过程中的团聚。AC经氧化改性后负载的Ru粒径更小,其中Ru/AC-H2O2中Ru的平均粒径为1.65 nm,而Ru/AC-HNO3中Ru的平均粒径为1.47 nm。这可能是由于Ru的前驱体在氧化改性后的载体表面分散较好,使得还原后能得到粒径较小的Ru。HRTEM结果显示,Ru在载体表面暴露的主要晶面为(101)和(100)。
2.6 XPS表征
还原后催化剂的XPS图谱见图6。
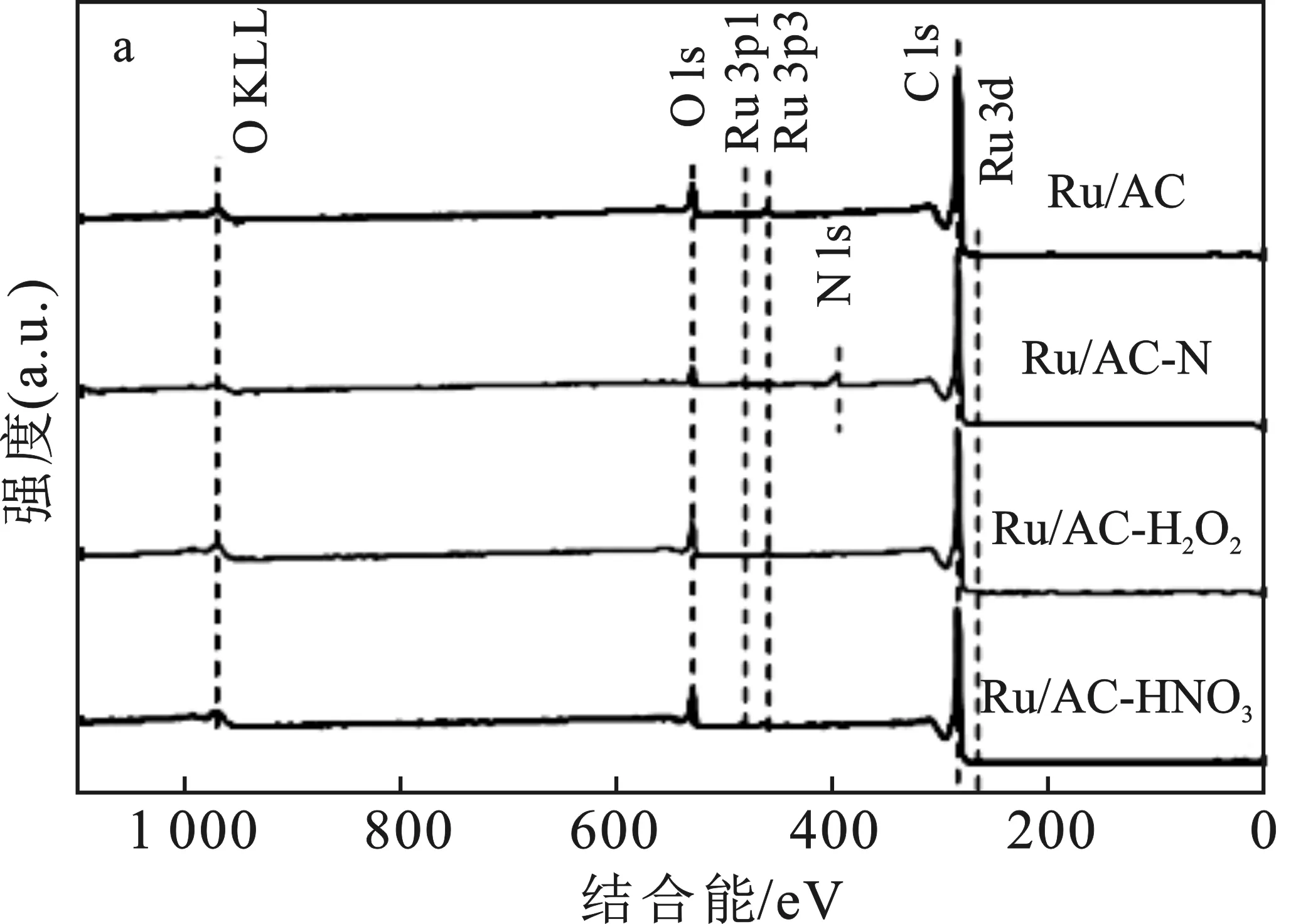


图6 催化剂的XPS谱图Fig.6 XPS spectra of the catalysts(a)全谱;(b)Ru 3d5谱;(c)N 1s谱;(d)O 1s谱
由图6可知,Ru 3d5的结合能均在280 eV左右,说明负载的Ru元素均被还原为Ru0。只有Ru/AC-N的XPS谱图中能观察到N的存在,Ru/AC-N上的N以吡咯氮和吡啶氮的形式共存,结合能400.5 eV处的峰归属于吡咯氮,结合能398.5 eV处的峰归属于吡啶氮。根据峰面积之比,吡咯氮占据了71.26%,吡啶氮占据了28.74%(见表2)。在这两种氮中,由于吡啶氮带有局域电子,可以向金属活性中心转移,常被认为是更重要的掺杂位点[18]。因此,Ru/AC-N中能和Ru形成相互作用的N的数量并不多。
由O 1s谱图可知,催化剂上的O以羟基氧和晶格氧的形式存在,结合能532 eV处的峰归属于羟基氧,结合能530 eV处的峰归属于晶格氧。对O 1s峰进行分峰拟合可以得出氧物种的比例及相对含量见表2。
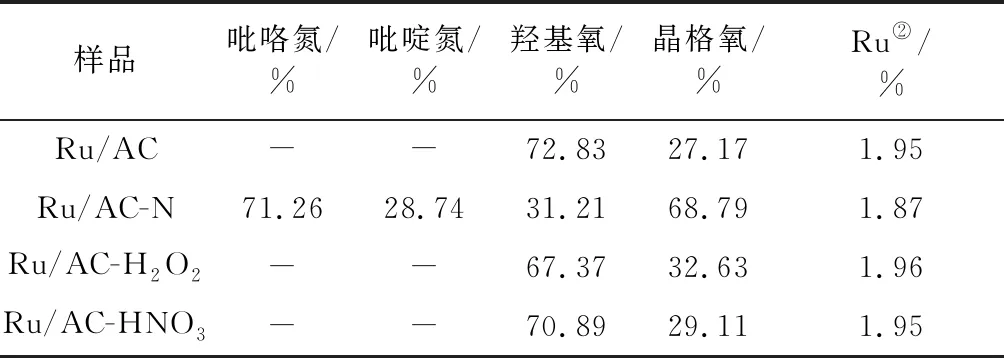
表2 催化剂表面物种比例①Table 2 Surface species ratios over the catalysts
由表2可知,氮掺杂之后,其表面氧的总含量明显减少。其中,羟基氧的比例减少,而晶格氧的比例增加。这是由于氮掺杂在高温条件下进行,会造成羟基氧的脱除,从而形成更多的晶格氧。另外,三聚氰胺也会与AC表面的羟基反应,造成羟基氧的减少。从表面氧含量来看,氧化改性之后,其表面的羟基氧和晶格氧数量均有所增加,强氧化改性的增量多于弱氧化改性。从相对比例来看,氧化改性后晶格氧所占的比例略有增加。该结果进一步证明,氧化改性在AC表面引入了含氧官能团。
2.7 载体表面对水的浸润性
通过分析载体在水中的分散情况来初步判断载体的亲疏水性。见图7,AC-H2O2和AC-HNO3在水中分散均匀,形成黑色的悬浊液。未改性的AC在水中的分散情况一般,溶液为灰色悬浊溶液,部分载体沉于溶液底部。AC-N在水中的分散最差,溶液颜色澄清透明,AC-N沉于溶液底部且和液体界限分明。

图7 样品在水中的分散情况Fig.7 Dispersion behavior of the samples in water(a)AC;(b)AC-N;(c)AC-H2O2;(d)AC-HNO3
因此,可初步断定,经过氧化改性后的载体样品AC-H2O2和AC-HNO3的亲水性较好,N掺杂改性的载体AC-N展现出明显的疏水性,而原始的AC载体的亲疏水性介于它们之间。
为了对载体的亲疏水性进行更准确的描述,通过双面胶将载体固定于载玻片表面,进行水的接触角测试,结果见图8。

图8 (a)AC,(b)AC-N,(c)AC-H2O2,(d)AC-HNO3,(e)玻璃以及(f)双面胶上水的接触角Fig.8 Water contact angle on(a) AC,(b) AC-N,(c) AC-H2O2,(d) AC-HNO3,(e) glass,(f) double-sided tape
由图8可知,与水在玻璃及双面胶上的接触角相比,4种载体对于水的接触角均有明显不同,说明载体颗粒覆盖满了双面胶表面,所测接触角为载体对水的真实接触角。AC对水的接触角为86.0°,<90°,属于亲水状态,但亲水性较弱。AC-N对水的接触角为141.2°,为疏水性材料,甚至接近超疏水状态(150°)。这是由于氮掺杂过程中,表面羟基大量损失,从而导致AC-N的疏水性大幅度升高。AC-H2O2对水的接触角为43.5°,弱氧化改性使得AC表面产生了更多的羟基亲水基团,因此其亲水性有明显的提升。AC-HNO3对水的接触角为21.9°,其亲水性最好。这是由于强氧化改性不仅产生了更多的羟基亲水基团,还生成了较多的羧基亲水基团,从而导致AC-HNO3的亲水性大幅度提升。
2.8 催化剂活性评价
催化剂的活性评价结果见图9。
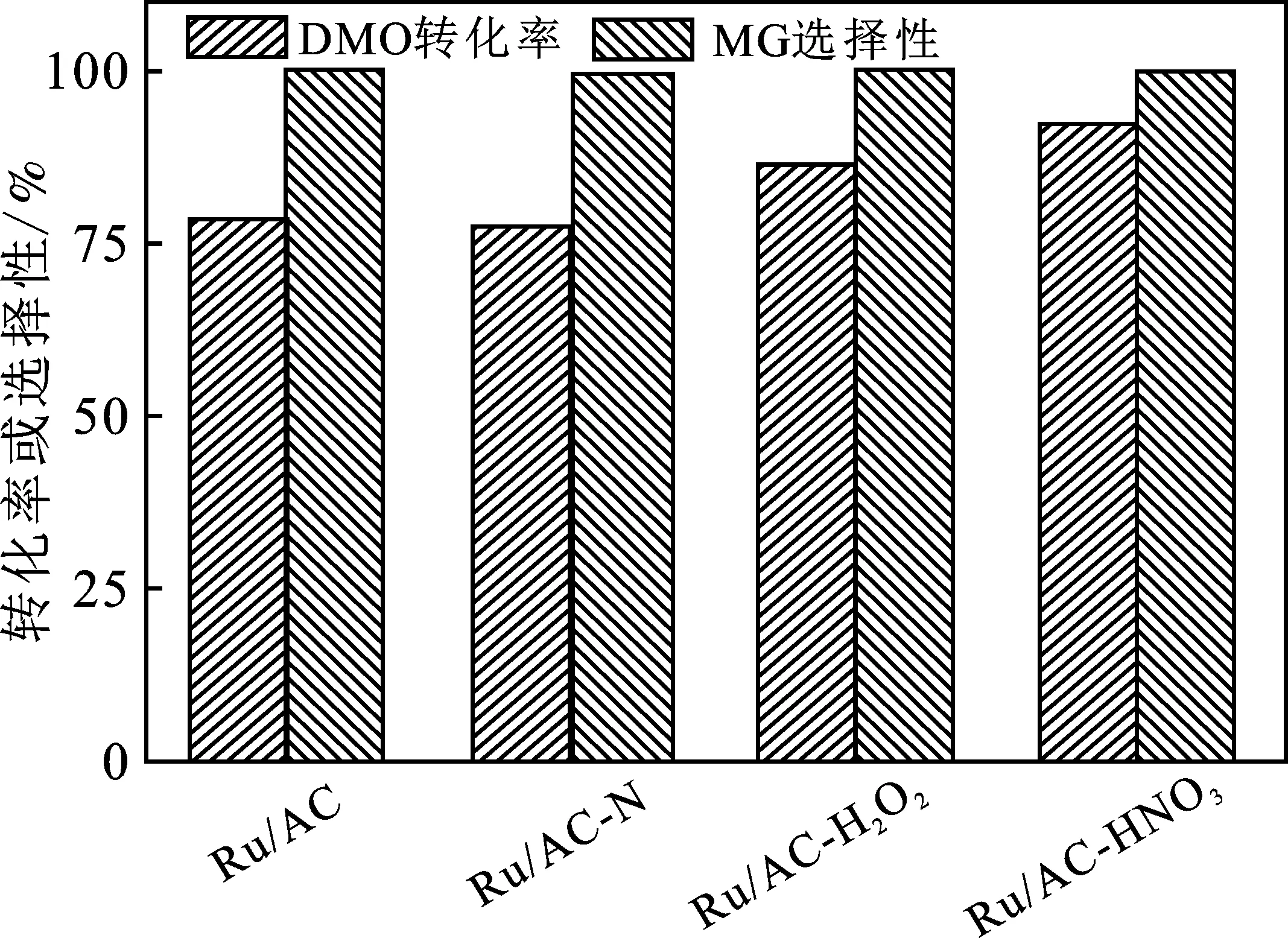
图9 催化剂的DMO加氢性能对比图Fig.9 Comparison of the catalytic performance over the catalysts
由图9可知,4种催化剂对于MG的选择性都接近100%,说明Ru基催化剂在低温下对于DMO加氢制MG具有很好的选择性。从转化率来看,Ru/AC和Ru/AC-N的DMO转化率比较接近,分别为78.3%和77.5%。这可能是因为Ru在载体表面的粒径大小接近,其活性位比表面也相差不大。虽然AC-N上的吡啶氮对Ru起着锚定作用,有利于Ru的分散,但是吡啶氮的含量较少,并且AC-N较大的疏水性对其分散有着不利的影响。因此,二者Ru的平均粒径相差不大。另外,氮掺杂对于载体的石墨化结构破坏较大,导致其导电性降低,这对Ru/AC-N的催化活性也是不利的。在Ru/AC-H2O2和Ru/AC-HNO3催化剂上,DMO的转化率明显提高,分别达到86.4%和92.3%。这与氧化改性提高了AC的亲水性,进而提升了Ru的分散度有关。同时,氧化改性对活性炭的石墨化结构破坏较小,有利于电子在载体和金属活性中心之间的传递。强氧化改性的Ru/AC-HNO3上Ru的平均粒径最小,其石墨化程度以及对水的浸润性都最好,因此Ru/AC-HNO3展现出最佳的加氢活性。
3 结论
采用氮掺杂、弱氧化以及强氧化的方法对活性炭载体进行表面改性用于制备Ru基催化剂。其中强氧化改性载体负载Ru的Ru/AC-HNO3催化剂展现出最佳的催化活性,其乙醇酸甲酯的收率达到了92.3%。氮掺杂改性由于N原子的引入,带来了更多的缺陷位,破坏了AC上的石墨化结构,且掺杂N主要以吡咯氮的形式存在,吡啶氮的含量较少,难以锚定Ru。同时,氮掺杂过程中造成了活性炭表面羟基的大量脱除,极大的降低了催化剂表面水的浸润性,不利于Ru的分散负载以及活性中心和反应物之间的接触。而强氧化改性引入了大量的羟基、羧基等亲水基团,提升了催化剂表面的水浸润性,有助于Ru前驱体溶液在载体表面的分散以及反应过程中催化剂与反应物的接触。此外,氧化改性还可维持碳载体表面的石墨化结构,有利于载体与活性中心之间的电子传递,进一步提高了催化加氢性能。
