基于高通量测序分析孤独症谱系障碍儿童肠道菌群的构成变化
2021-03-09曾佩佩邓梁琼冯玉山黄丽萍李红辉
曾佩佩, 曾 婷, 邓梁琼, 郭 浩, 冯玉山, 黄丽萍, 李红辉
(1.柳州市妇幼保健院,广西 柳州 545001;2.北京量化健康科技有限公司,北京 100070)
孤独症谱系障碍(autism spectrum disorder,ASD)属于广泛性发育障碍中的一个类型,主要表现为社会交往障碍、言语和非言语交流障碍、刻板行为和狭隘兴趣三大主要临床症状[1]。近年来,ASD患病率呈现逐年上升的趋势。BAIO等[2]的研究结果显示,美国8岁儿童中每59人就有1人患有ASD,男女患病比例为4∶1。我国尚无大样本流行病学报道。目前,ASD的干预方法以行为治疗为主,无法达到满意的康复效果。ASD的发病机制尚不明确,近年来随着对肠道微生物研究的不断深入,有学者发现肠-脑-微生态轴的紊乱可能与ASD的发生、发展有重要关系[3]。本研究拟采用高通量测序分析ASD患儿粪便样本的肠道菌群分布,以期为ASD患儿的肠道微生态研究提供依据。
1 材料和方法
1.1 研究对象
选取2019年2—3月柳州市妇幼保健院儿童保健科确诊为ASD的患儿35例(ASD组),其中男28例、女7例,年龄(3.11±0.83)岁。选取同期柳州市妇幼保健院体检健康儿童35名(正常对照组),其中男21名、女14名,年龄(3.37±1.24)岁。2个组之间年龄、性别差异均无统计学意义(P>0.05)。本研究经柳州市妇幼保健院伦理委员会批准,所有对象的家属均签署知情同意书。
1.2 纳入与排除标准
ASD患儿均符合美国精神障碍诊断和统计手册第5版(Diagnostic and Statistical Manual of Mental Disorders -fifth edition,DSM-Ⅴ)规定的ASD诊断标准。年龄2~6岁,性别不限。排除患有重大疾病者,如糖尿病、结肠炎、痢疾、肝硬化等。近1个月内未使用过抗菌药物、激素类药物以及注射过病毒类疫苗,未服用益生菌及益生元补充品。自愿参加本研究,且家属签署知情同意书。
1.3 方法
1.3.1 粪便标本采集 粪便标本采集采取立取、立存(取样后立即保存)原则,用专用无菌粪便采集盒留取粪便,排便之前排空小便,防止样本接触尿液,影响检测结果。取样前洗手,戴无菌手套、口罩,用取便管深入粪便,采集约2 g粪便置于样本保护液中保存。样本采集后,标记好姓名、标号、采样日期等以备检查。所有对象均需完整、详细填写基本信息调查表,经核对准确无误后建立完整资料数据库。
1.3.2 样本DNA提取、文库构建及测序 对所有粪便样本进行DNA提取并构建文库。采用nova seq 6000测序仪(美国Illumina公司)及配套试剂进行测序,得到原始测序数据。所有样本均进行生物信息数据分析,得出肠道菌群信息,并确定菌群特征。
1.3.3 生物信息数据分析 原始宏基因组测序数据采用MOCAT2软件进行质量控制[4]。高质量的reads用SOAPdenovo v2.04软件(参数:all-D 1-M 3-L 500)进行从头组装,基于组装获得scaftigs,用MetaGeneMark软件进行基因结构预测[5],预测后的基因用CD-HIT聚类算法进行聚类去冗余[6],构建基因长度>100 bp的非冗余基因集。采用DIAMON软件将获得的非冗余参考基因集与京都基因组百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)和碳水化合物活性酶数据库(Carbohydrate-active enzymes database,CAZy)比对,对预测出的基因进行功能注释。在基因层面上,将高质量的reads通过BWA软件与构建的非冗余基因集进行对比,去除长度<30 bp及一致性<95%的reads,得到每个基因的reads计数,再经标准化处理获得基因的相对丰度。在菌群上进行样本内多样性分析,用Shannon指数表示样本的alpha多样性,Shannon值越大,说明群落多样性越高。用基于bray-curtis 距离的主坐标分析(principal coordinate analysis,PCoA)在二维坐标上显示不同样本之间的相似性及差异性。采用相似百分比方法确定造成菌群群落差异的主要细菌种类[7]。用PERMANOVA分析[8]检验分组对样本菌群差异的影响。通过Wilcoxon检验比较组间菌群相对丰度的差异,以确定有显著差异的单元。采用Lefse分析寻找在各分类层级上差异显著的单元。
1.4 统计学方法
采用SPSS 19.0软件进行统计分析。呈正态分布的数据以±s表示,2个组之间比较采用两独立样本t检验,以P<0.05为差异有统计学意义。
2 结果
2.1 测序结果统计
35例ASD患儿和35名正常对照者测序后共得到408.3 G原始测序数据,样本的reads为23 494 234~69 105 708,平均为41 748 599。测序结果进行质量控制后再进行从头组装、基因结构预测、聚类去冗余,最终获得非冗余基因集。2个组之间基因的韦恩图和box图见图1。韦恩图提示ASD组与正常对照组粪便样本中基因表达存在差异。box图显示2个组粪便样本基因的集中性均较好,无明显异常值。ASD组粪便样本基因表达较正常对照组更集中。

图1 ASD组与正常对照组粪便样本中基因表达的韦恩图和box图
2.2 菌群组成分析
对所有研究对象的肠道菌群从物种到门水平各个层级进行分析,获得相对丰度。结果显示,ASD组和正常对照组的肠道菌群共包含16个门159个属,其中糖酵母菌门2个组之间差异有统计学意义(P=0.01),但其在肠道菌群结构中相对丰度很低;厚壁菌门在所有菌群中占最主要地位,所有研究对象的优势菌门依次为厚壁菌门、拟杆菌门、变形菌门、放线菌门,2个组之间优势菌门的相对丰度差异均无统计学意义(P>0.05)。ASD组与正常对照组之间罕见小球菌属、霍尔德曼菌属、Candidatus Saccharibacteria noname属、普雷沃菌属、Burkholderiales noname属、戈登杆菌属、另枝菌属、Solobacterium属、Parasutterella属、Anaerotruncus属、杆菌属、丛毛单胞菌属、孪生球菌属、颗粒球菌属相对丰度差异均有统计学意义(P<0.05),但除另枝菌属外,其他菌属相对丰度都很低。相对丰度居前8位的分别为拟杆菌属、双歧杆菌属、粪杆菌属、优杆菌属、Blautia属、另枝菌属、梭菌属及瘤胃球菌属。ASD组与正常对照组之间仅Alistipes属相对丰度差异有统计学意义(P=0.022)(ASD组占6.34%,正常对照组占2.44%),其他7个菌属2个组之间差异均无统计学意义(P>0.05),见表1。ASD组与正常对照组各菌门和菌属的相对丰度比例见图2。ASD组与正常对照组菌群多样性分析的Shannon指数见图3。基于菌群计算的样本距离PCoA结果显示,主坐标成分为12.5%,即样本菌群存在差异,见图4。造成菌群差异的主要细菌种类前20位分别为长双歧杆菌、直肠真杆菌、Faecalibacterium prausnitzii、脆弱拟杆菌、粪普雷沃菌、普通拟杆菌、短双歧杆菌、单形拟杆菌、Alistipes putredinis、Faecalibacterium prausnitzii、居粪拟杆菌、Ruminococcus bromii、多形拟杆菌、椭圆形拟杆菌、假小链双歧杆菌、活泼瘤胃球菌、Clostridium nexile、鼠乳杆菌、Eubacterium eligens及粪拟杆菌。Permanova检验结果显示分组对样本菌群差异无影响(P=0.178),见图5。


图2 不同分组在菌门和菌属水平的相对丰度组成

图3 ASD组与正常对照组菌群多样性的比较

图4 基于菌群计算的样本距离PCoA结果
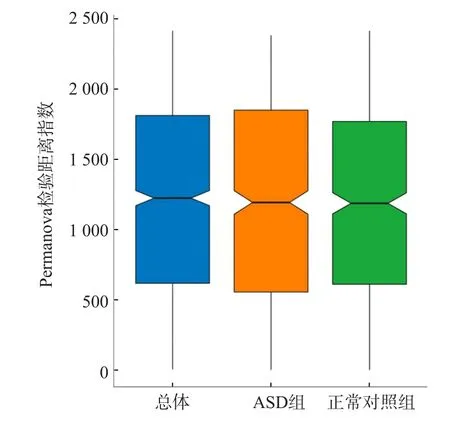
图5 Permanova检验结果
2.3 菌群差异分析
组间菌群相对丰度的差异分析结果显示,ASD组翁德顿氏另枝菌、Alistipes putredinis、芬戈尔德氏拟杆菌、谢氏另枝菌、前庭链球菌、丝状霍尔德曼菌、芽胞拟杆菌、伯克霍尔德菌-1-1-47、单形巨单胞菌、Gordonibacter pamelaeae、粪副拟杆菌、生黄瘤胃球菌、毛螺科菌_3_1_57FAA_CT1、Parasutterella excrementihominis的相对丰度明显高于正常对照组(P<0.05),而毗邻颗粒链球菌、死亡梭杆菌、莫氏细小杆菌、直肠真杆菌、Candidate division_TM7_single_cell_isolate_TM7c及麻疹孪生球菌相对丰度低于正常对照组(P<0.05),见表2。

表2 ASD组与健康对照组之间前20位菌群相对丰度差异比较
Lefse分析结果显示,ASD组与正常对照组菌群在各分类层级上均存在有显著差异的单元,其中ASD组粪便样本中显著存在的是普雷沃菌科、理研菌科、Burkholderiales noname科及丛毛单胞菌科,而正常对照组粪便样本中显著存在的是肉杆菌科、梭菌科、氨基酸球菌科及伯克氏菌科。见图6。

图6 Lefse分析结果
2.5 菌群差异功能分析
针对基于基因加和而成的KEGG和CAZy功能层面的read count丰度,分析ASD组与正常对照组的差异。ASD组与正常对照组注释到KEGG层面及CAZy层面的基因相对丰度差异有统计学意义(P<0.05)。差异居前20位的基因的身份识别号(identity document,ID)见表3、表4。

表3 ASD组与正常对照组注释到KEGG层面的基因丰度差异居前20位基因的ID

表4 ASD组与正常对照组注释到CAZy层面的基因丰度差异居前20位基因的ID
3 讨论
肠道菌群是人体肠道的正常微生物,定居于肠道黏膜表面,能产生名为N-酰基酰胺的小型有机复合物,可与受体相互作用,调节人体生理健康,参与包括免疫、行为和代谢等各个方面的信号传导[9]。肠道菌群与宿主相互影响的体系统称为肠道微生态,是人体最大的微生态系统,该系统通过神经、内分泌免疫及代谢途径影响中枢神经及肠道系统,这种相互作用即为肠-脑轴-微生态[10]。肠道微生物菌群对人类儿童阶段的生长发育起重要作用[11]。近期有研究结果显示,ASD的发病与肠道菌群的紊乱相关[12-13],ASD患者的肠道菌群构成与健康人群存在差异。CORETTI等[14]采用16SrRNA测序并扩增16SrRNA(V3~V4)基因标记对2~4岁ASD患儿及同年龄健康儿童肠道菌群进行对比,结果显示,ASD患儿拟杆菌门和变形杆菌门明显增多,放线菌门明显减少。STRATI等[15]通过焦磷酸测序分析发现ASD患者中厚壁菌门/拟杆菌门比值显著升高,在属水平上,ASD组另枝菌属、嗜胆菌属、小杆菌属、副拟杆菌属和韦荣球菌属的相对丰度下降,而柯林斯氏菌、棒状杆菌、Dorea和乳酸菌的相对丰度明显上升。MARÍNEZ-GONZÁLEZ等[16]通过回顾性分析发现,ASD患儿念珠菌、普雷沃菌、链球菌和细孔菌的丰度高于有便秘症状的儿童(P<0.05),二者之间拟杆菌门、厚壁菌门、放线菌门、杜氏菌门、乳酸菌门、Prausnitzii、粪杆菌门及拟杆菌门/厚壁菌门比值有明显差异。ZHANG等[17]采用16SrRNA测序并扩增16SrRNA(V3~V4)基因标记对35例ASD患儿的肠道菌群进行研究,结果显示,在门水平上,ASD组拟杆菌门/厚壁菌门比值显著升高;在属水平上,ASD组萨特菌属、Odoribacter属和Butyricimonas属的基因相对丰度显著高于正常对照组(P<0.05),而韦荣球菌属和链球菌属的基因相对丰度显著低于正常对照组(P<0.05)。MA等[18]对45例ASD患儿与45名健康儿童的粪便样本进行16SrRNA测序发现,ASD患儿肠道菌群在门水平上的微生物群相对丰度与健康儿童相比差异无统计学意义(P>0.05),在其他层面上,ASD患儿与健康儿童相比,氨基酸球菌科相对丰度较低,Lachnoclostridium属、Tyzzerella亚群4、Flavonifractor属和Lachnospiraceae属的相对丰度降低[18]。目前,ASD患者肠道菌群失调已被证实,但不同的患者微生物群改变并不一致,而且常常相互矛盾,并没有一个独特的微生物群组成[19]。
在菌门水平上,健康人群肠道菌群中存在厚壁菌门、变形菌门、放线菌门及拟杆菌门,这些细菌在肠道内形成一定的比例,从而控制着肠道菌群的平衡,这种关系可以直接反映宿主本身的健康状况。本研究结果显示,ASD患儿肠道菌群中存在4种优势菌门——厚壁菌门、拟杆菌门、变形菌门及放线菌门,与正常对照者比较差异均无统计学意义(P>0.05)。本研究结果与国外研究结果[14,16]存在差异,与国内部分研究结果[18]一致,这可能与地域差异、饮食结构差异有关。本研究结果还显示,ASD组与正常对照组糖酵母菌门差异有统计学意义(P=0.01),但其在肠道菌群结构中的相对丰度很低。目前,对人肠道系统中糖酵母菌门的研究较少,其主要被发现存在于活性污泥中,用于处理工业污水[20]。因此,糖酵母菌门是否与ASD发病机制相关仍需进一步研究来证实。
本研究结果显示,在菌属水平上,ASD组与正常对照组有14种菌属存在差异,其中相对丰度较高的另枝菌属在文献[15]中有过报道,其余13种菌属相对丰度均很低,在既往的研究中鲜有报道,因此其与ASD的相关性尚需进一步深入研究。另枝菌属属于理研菌科,是革兰阴性厌氧菌,为兼性致病菌。有研究结果显示,抑郁症患者肠道系统中另枝菌属增多[21-22],可能是导致抑郁症的主要原因之一[23]。另枝菌属可影响色氨酸的利用,破坏肠道系统中5-羟色胺的平衡,进而抑制大脑活动,这可能是导致神经系统障碍的原因之一[24],但其与ASD的相关性仍有待进一步研究加以证实。
本研究菌群差异分析结果显示,ASD组翁德顿氏另枝菌、Alistipes putredinis、芬戈尔德氏拟杆菌、谢氏另枝菌、前庭链球菌、丝状霍尔德曼菌、芽胞拟杆菌、伯克霍尔德菌-1-1-47、单形巨单胞菌、粪副拟杆菌、生黄瘤胃球菌、毛螺科菌_3_1_57FAA_CT1及Parasutterella excrementihominis的相对丰度均高于正常对照组(P<0.05),而毗邻颗粒链球菌、死亡梭杆菌、莫氏细小杆菌、直肠真杆菌、Candidate division_TM7_single_cell_isolate_TM7c及麻疹孪生球菌相对丰度均低于正常对照组(P<0.05)。Lefse分析结果显示,ASD组粪便样本中显著存在的是普雷沃菌科、理研菌科、Burkholderiales noname科及丛毛单胞菌科,正常对照组粪便样本中显著存在的是肉杆菌科、梭菌科、氨基酸球菌科及伯克氏菌科。进一步利用KEGG及CAZy对差异菌群功能进行注释,结果显示注释到的基因相对丰度在ASD组与正常对照组之间存在明显差异,提示碳水化合物活性酶可能参与了ASD的发病过程。
综上所述,ASD患儿肠道存在微生态失调,与既往研究结果[14-18]一致,但检出的细菌种类与既往研究结果[14-18]存在差异,进一步验证结果显示ASD患儿并没有一个特定的肠道微生物群组成[19]。本研究检测出的细菌有可能是ASD发生、发展的潜在生物学标志物。但本研究尚存在一定的局限性:纳入样本量不足,研究对象年龄集中在2~6岁,且探讨的是我国儿童的肠道菌群情况,与国外研究相比,研究对象的生活环境不同、饮食结构有一定的差异,因此对研究结果有一定影响。今后将扩大样本量,进行更大范围的研究,为ASD患儿肠道微生态研究提供更充分的依据。
