Sotos综合征患儿临床特征及基因变异分析
2021-03-09郑洪雪殷丽萍王秀敏
郑洪雪, 陈 瑶, 殷丽萍, 李 辛, 丁 宇, 李 娟, 王秀敏
(1.常州市第一人民医院 苏州大学附属第三医院,江苏 常州 213000;2.上海交通大学医学院附属上海儿童医学中心内分泌代谢科,上海 200127)
Sotos 综合征是一种常染色体显性遗传的过度生长性疾病,人群发病率约为1∶14 000[1],其诊断标准为特殊面容(额头突起、眼裂下斜、眼距过宽、下颌尖长、高腭弓、 双颞部毛发退化等)、过度生长(头围增大、身高增高,且大于正常同龄儿童的第97百分位数)、骨龄超前、发育迟缓(语言和学习障碍、短期记忆和抽象思维能力缺陷,可伴有不同程度的智力低下等);此外,还可有早期喂养困难、黄疸、肌张力低下、动作笨拙、协调性差及癫痫等非特异性表现[2]。Sotos综合征一般以生长过快为主要表现,骨龄明显超前,临床如对该病认识不足,易与性早熟混淆。本研究拟分析1例误诊为性早熟的Sotos综合征的临床特征及基因突变特点。
1 材料和方法
1.1 病例介绍
患儿,男,6岁11个月,因“生长过快5年余”于2019年1月10日就诊,具体生长速度不详。患儿系第1胎第1产,试管婴儿,足月剖宫产,出生体质量3 400 g,出生身长不详,无窒息抢救病史。出生后有反复黄疸病史,无喂养困难,有新生儿肺部感染病史,2岁时诊断脑积水(静止性),有热性惊厥病史,行视频脑电图及动态脑电图均未见异常,头颅磁共振成像(magnetic resonance imaging,MRI)提示幕上脑室增宽,双颞极颅板下、左侧小脑左侧脑外间隙增宽,左侧椎动脉明显变细。心脏彩色多普勒超声检查示左心房、左心室明显增大,左心室侧壁心肌组织疏松,二尖瓣轻中度反流,左心收缩功能正常。心脏MRI提示左心室心尖及侧壁心肌较疏松,建议MRI随访。抬头、翻身均较同龄人晚,15个月会独走,近20个月时会说话,智力发育较同龄人略落后。有频繁呼吸道感染病史。父亲身高180 cm,母亲身高160 cm,祖父身高170 cm,祖母身高155 cm,父母非近亲婚配,其他家族成员无类似病史。就诊前因“ 社区获得性肺炎,非重症”在上海交通大学医学院附属上海儿童医学中心呼吸科住院治疗,好转出院。住院期间先后予“红霉素、头孢呋辛、头孢唑肟、甲基强的松龙、米诺环素”治疗。
1.2 方法
在征得患儿家长知情同意,并经上海儿童医学中心伦理委员会批准后,对患儿及其父母进行基因检测。采集患儿及其父母血液样本各2 mL。采用QIAamp DNA Blood Mini试剂盒(德国Qiagen GmbH公司)提取外周血基因组DNA。基因组DNA经打断处理后得到150~200 bp的DNA片段,采用Sure Select Inherited Diseases Test(2 724 genes)试剂盒建库(美国Agilent公司),采用Illumina测序平台(美国Illumina公司)进行高通量测序。测序数据经Next GENe软件(美国SoftGenetics公司)匹配分析后,采用Ingenuity Variant Analysis软件(美国Ingenuity Systems 公司)进行生物信息学分析。
2 结果
2.1 外院检查结果
本例患儿就诊前15个月因生长过快,在当地医院行骨龄检查,提示骨龄6~7岁(家属未提供骨龄X线片)。性激素激发试验结果显示,促黄体生成素(luteinizing hormone,LH)峰值为8.45 mIU/mL,促卵泡刺激素(follicle-stimulating hormone,FSH)峰值为6.91 mIU/mL。睾丸B超结果显示,右侧睾丸19 mm×9 mm,左侧睾丸19 mm×9 mm。睾酮<0.087 nmol/L,人绒毛膜促性腺激素<0.5 mIU/mL,染色体46XY,查体未见阴毛。诊断为性早熟。给予醋酸曲普瑞林治疗15个月。
2.2 就诊后的查体结果
患儿身高为148.3 cm[高于同年龄、同性别、同地区儿童平均身高3个标准差(+3s)],近15个月身高增长10 cm;体质量为47.3 kg[高于同年龄、同性别、同地区儿童平均体质量3个标准差(+3s)]。特殊面容:头围58 cm、前额突出、下颌窄、高腭弓,见图1。外生殖器性成熟分级为Tanner Ⅰ期,睾丸G1期,阴茎长度3 cm,阴毛PH1,语言沟通尚可,智力较同龄人稍落后,运动协调能力稍差。骨龄为8~9岁,X线片示各掌指骨形态正常,骨质结构无破坏,见图2。患儿治疗期间的生长曲线图示生长速度未见减缓,见图3。

图1 本例患儿特殊面容

图2 本例患儿2019年1月10日的左腕正位片

图3 患儿治疗前、治疗中及治疗后的生长曲线
2.3 基因检测结果
患儿核受体结合SET域蛋白1(nuclear receptor-binding SET-domain-containing protein1,NSD1)基因(NM_022455.4)存在“错义变异c.5854C>T(p.Arg1952Trp)(杂合)”,与目前症状相关的其他基因(IGF2、H19、CDKN1C、KCNQ10T1)未发现病理性变异,患儿父亲携带该变异位点(杂合),见图4。经Alamut功能软件预测可能会影响蛋白结构域的功能,按照美国医学遗传学和基因组学学会(American College of Medical Genetics and Genomics,ACMG)变异分类标准,归类为“可能致病性变异”。
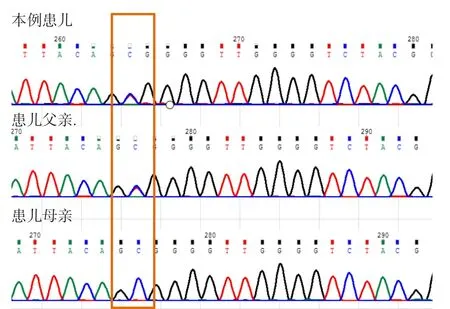
图4 NSD1基因测序图
3 讨论
NSD1基因定位于5号染色体q35位置,其编码的蛋白可以直接与核受体相互作用,发挥激活或阻遏转录的作用。有70%~90%的Sotos综合征患者存在NSD1基因突变,其中多数患者是点突变,约有10%为5q35NSD1基因微缺失[3]。NSD1基因包含23个外显子,其中2~23外显子为编码区;cDNA长度为8 552 bp,包含1个8 088 bp的开放阅读框,编码含有2 696个氨基酸的蛋白质。此蛋白至少有10个功能性结构域(NID-L、NID+L、SET、PWWP-Ⅰ、PWWP-Ⅱ、 PHD-Ⅰ、PHD-Ⅱ、PHD-Ⅲ、PHD-Ⅳ和PHD-Ⅴ)[4-5]。NSD1编码的蛋白是一种独特的具有2种不同功能的调节辅助因子,可以表达于大脑、骨骼肌、肾脏、脾脏、胸腺、胎盘、肺和外周血白细胞中[6],作为生长基因辅阻遏物发挥作用[7]。
Sotos综合征是由NSD1基因缺失或突变而导致的单倍体剂量不足(单倍体剂量不足指一个等位基因突变后,另一个等位基因能正常表达,编码的蛋白质只有正常水平的50%,不足以维持细胞正常的生理功能)引起的。大多数Sotos综合征患者有轻到中度的智力障碍,这与NSD1基因的突变类型有关。同时,有30%~50%的Sotos综合征患者有癫痫发作史,其中50%的患者有发热性癫痫发作[2,8-9]。本例患儿2岁时有热性惊厥病史,但多次行脑电图检查均未见异常,需进行随访以观察病情变化。NSD1基因突变和染色体微缺失的患者均会出现智力障碍、儿童期过度生长等症状,但与基因突变患者相比,染色体微缺失的患者智力障碍程度重、过度生长表现轻[10]。本例患儿为NSD1基因错义突变,存在轻度智力发育落后,其父亲携带该位点变异,但父亲仅表现为身材高大,无其他临床病理特征,提示NSD1基因存在差异性表达的可能,具体机制尚不明确;患儿祖父、祖母无相应临床表现,因个人原因不愿意完善基因测序验证,因此尚不能明确患儿父亲突变位点的具体来源。
过度生长性疾病一般会导致肿瘤的发病率升高,但Sotos综合征患者的肿瘤发病率<3%[5,11]。近年来,已有神经母细胞瘤中NSD1基因表观遗传失活的报道,研究者认为NSD1的失活甚至对神经母细胞瘤的预后判断也有一定价值[12]。鉴于这种一致的罕见性和新发现的遗传联系,需要进一步的研究来评估这种相关性及在相关疾病预后判断中的意义。COLE等[2]的研究结果显示,至少有15%的Sotos综合征患者存在骨龄超前、新生儿黄疸、婴儿期喂养困难、脑发育异常、肌张力低、脊柱侧弯、心脏及肾脏异常、关节过度松弛、扁平足等临床特征。本例患儿有新生儿黄疸、脑发育异常、骨龄超前;心脏彩色多普勒超声检查结果提示左心房和左心室明显增大,左心室侧壁心肌组织疏松,二尖瓣轻中度反流,左心收缩功能正常,与文献报道[13]相符。另外,日本学者NAKAMURA等[14]报道了1例10岁男性Sotos综合征患儿,双孔二尖瓣合并左心室心肌致密化不全。神经影像学报告提示Sotos综合征患儿脑室扩大,中线结构异常(胼胝体发育不全、小脑腔、脑萎缩)高发[15]。本例患儿头颅MRI结果提示,幕上脑室增宽,双颞极颅板下、左侧小脑左侧脑外间隙增宽,左侧椎动脉明显变细,与文献报道[16]相符。
新生儿Sotos综合征有短暂的高胰岛素血症性低血糖(hyperinsulinemic hypoglycemia,HI),这虽为Sotos综合征不常见的临床表现,但NSD1基因单倍体剂量不足足以引起新生儿HI,因此新生儿HI患儿应考虑Sotos综合征,这也有助于扩展Sotos综合征的表型谱[17]。关于Sotos综合征患者皮肤问题的报道较少,曾有短暂性新生儿皮肤松弛症和多毛症作为Sotos综合征首发症状的报道[18]。LACCETTA等[19]报道了3例1家系3代NSD1错义突变导致的Sotos综合征,其中先证者母亲及外祖父仅表现为孤立性生长,提示NSD1基因突变是否直接影响生殖健康尚不明确;表观遗传机制和子宫内环境可能是基因变异差异性表达的影响因素,因此基因检测不能完全预测表型。
本例患儿因过度生长在当地医院就诊,行性激素激发试验,LH峰值为8.45 mIU/mL,FSH峰值为6.91 mIU/mL,骨龄为6~7岁,诊断为性早熟。但患儿查体结果示外生殖器性成熟分级为TannerⅠ期,阴毛PH1期,阴茎长度3 cm,性腺无增大,仅凭生长过快、骨龄超前及性激素激发试验结果无法诊断为性早熟,可能与该病临床少见,临床医生对该病认识不足有关,提示需进一步完善基因检测以明确诊断。患儿治疗前后生长曲线显示经醋酸曲普瑞林治疗15个月后,生长速度未见减缓。WIT等[20]研究了22例Sotos综合征患儿,结果显示所有患儿均存在骨龄超前,4岁之前患儿的骨龄超前1.0~2.3岁不等,5岁以后骨龄与实际年龄的差异稳定在2.0~2.8岁。本例患儿骨龄与实际年龄之间的差异与文献报道[20]相符。Sotos综合征患儿虽然骨龄提前,但青春期的开始往往是正常的,说明Sotos患儿骨龄提前可能与性激素水平关系不大。Sotos患儿骨龄显著提前,可能导致患儿提前达到成年终身高。另外,关于Sotos综合征患者性激素方面的研究甚少,传统的性激素激发试验中LH峰值>5 mIU/mL是否可以作为判断Sotos综合征患者青春期启动的依据还有待进一步探讨。
另外,还有多种疾病,如Weaver综合征(具有长头巨脑、精神发育迟缓、特异面容和四肢形态异常等临床特征)、Bannayan-Riley-Ruvalcaba综合征(主要特征为巨头、生殖器着色斑病和肠息肉病)、Beckwith-Wiedemann综合征(主要特征为脐膨出、巨舌和巨体,还可伴有其他畸形和异常,良性家族性巨颅仅表现为头颅巨大畸形,无其他脏器畸形表现)与Sotos综合征有相似的临床表现,极易混淆,通过后期随访及基因检测可进行鉴别。
Sotos综合征目前无特效治疗方法,以对症治疗为主。新生儿期黄疸可采用光疗等方法进行治疗,发育迟缓患儿可进行早期康复治疗,智力障碍患儿可进行特殊教育,同时应注重随访。随访过程中应进行生命体征各项指标评估,尤其是对于心脏、脊柱、脑部影像学检查有异常的患儿,随访间隔时间应相应缩短。如有症状者应及时就诊,尽早干预,以改善患儿生活质量及预后。
