LincRNA-EPS对脂多糖诱导的小鼠牙周膜细胞炎症因子表达的影响
2021-03-07叶远舟王雅冰苏俭生
叶远舟, 刘 凯, 王雅冰, 苏俭生
(上海牙组织修复与再生工程技术研究中心,同济大学口腔医学院,同济大学附属口腔医院口腔修复科,上海 200072)
牙周炎是一种病因及成因复杂的感染性疾病[1],也是最常见的口腔疾病之一。有研究显示,2010年全球重度牙周炎的患病率高达10.8%,影响约7.43亿人,是全球第六大流行疾病[2]。牙周炎的发病机制被认为是高度复杂的,由Toll样受体识别微生物配体激活的信号转导途径,引发了牙周炎症的产生[3-4]。牙周炎致病菌(革兰氏阴性杆菌)细胞外膜上的脂多糖(LPS)可被 Toll样受体 4(Toll-like receptor 4,TLR4)识别,从而介导免疫炎症的发生,在牙周炎的发生、发展中起重要作用[5-6]。炎症性免疫反应被认为是宿主防御感染的防御机制,但炎症性细胞因子的释放会导致牙龈萎缩和牙槽骨吸收,最终可导致牙齿脱落,严重影响口腔健康[7]。长链非编码 RNA(long noncoding RNAs,lncRNAs)是一类内源性长度大于200 nt的转录本,被认为一般不具有转录活性,因其在基因调控中的重要作用而成为近年来的研究热点。在免疫炎症过程中,lncRNAs同样发挥了重要作用[8-9]。Atianand等[10]在Cell杂志上发表了新的发现,lincRNA-EPS可通过控制核小体定位抑制免疫反应基因(immune response genes,IRGs)的转录,从而下调IRGs的表达。而LPS的刺激可以激活TLR4下调lincRNA-EPS的表达,导致免疫炎症反应的增强。那么lincRNA-EPS在LPS诱导的牙周炎症中是否发挥作用,以及发挥了怎样的作用,目前尚缺乏研究。本研究旨在初步探索lincRNA-EPS对LPS诱导的小鼠牙周膜细胞(mPDLCs)炎症因子表达的影响,并探讨其与牙周炎发生、发展过程中可能存在的内在联系。
1 材料和方法
1.1 材料
1.1.1实验动物 6~8周龄C57BL/6雄性小鼠(同济大学实验动物中心提供);6~8周龄lincRNA-EPS敲除的C57BL/6雄性小鼠(上海南方模式生物科技股份有限公司合作构建)。本实验经同济大学附属口腔医学院伦理委员会批准后进行,审批编号为[2018]伦审字(026)号。
1.1.2实验试剂 大肠埃希菌来源的LPS、Ⅰ型胶原酶(Sigma-Aldrich 公司,美国);胎牛血清,α-MEM 培养液(Gibco 公司,美国);波形蛋白(Vimentin,武汉三鹰生物技术有限公司,中国);荧光(Cy3)标记羊抗兔IgG(武汉博士德生物工程有限公司,中国);TRIzol试剂,Lipofectamine转染试剂盒(Invitrogen公司,美国);胰蛋白酶,青链霉素溶液(Hyclone公司,美国);lincRNA-EPS过表达质粒(上海吉凯基因化学技术有限公司,中国);逆转录试剂盒(TaKaRa公司,日本);ELISA 试剂盒:cxcl10(Invitrogen 公司,美国),IL-1α、IL-6(Novus公司,美国)。
1.1.3实验器材 CO2细胞培养箱 (Thermo公司,美国);Ⅰ型超净工作台(苏州净化设备有限公司,中国);体式显微镜、荧光倒置相差显微镜和照相系统(Zeiss公司,德国);低温高速离心机(大龙兴创实验仪器有限公司,中国);逆转录仪(Bio-Rad公司,美国);紫外线分光光度仪(GE公司,美国);荧光定量PCR仪 (Roche公司,瑞士);多功能酶标仪(BioTek 公司,美国)。
1.2 方法
1.2.1lincRNA-EPS基因敲除小鼠模型的构建 体外转录合成Cas9 mRNA和gRNA,用2%琼脂糖凝胶电泳检测。将20 μL 25 ng/mL的gRNA和10 μL 100 ng/μL的Cas9 mRNA混匀,加入3 mol/L乙酸钠3 μL,pH调至5.2,用无水乙醇沉淀后,再用无酶水重悬。通过显微注射法将混合物注射到小鼠受精卵胞质中,将存活的受精卵移植到假孕受体母鼠体内,至母鼠生产,获得首建鼠(F0代)。F0代小鼠PCR产物经T-vector连接后测序,比对测序结果。
1.2.2mPDLCs分离培养及鉴定 将小鼠断颈处死后,用75%乙醇浸泡5 min,随后将其转移至超净工作台内。用仰卧位固定小鼠,打开其口腔,用止血钳拔下完整的磨牙后,在体视显微镜下清除周边多余的牙龈组织及黏附的牙槽骨。用含4%青链霉素的磷酸盐缓冲液 (phosphate buffered saline,PBS)冲洗3次,加入2 mg/mL的Ⅰ型胶原酶1 mL,经37℃水浴摇床消化1 h后终止酶消化,用吸管反复吹打数次,消化液经200目细胞筛过滤,滤液经300×g离心5 min后,弃上清液,保留细胞沉淀物。加入含10%胎牛血清及1%青链霉素的α-MEM细胞培养液重悬细胞,接种于培养瓶中,于37℃、5%CO2恒温培养箱中静置培养,48 h后更换新鲜完全培养液,之后每3 d换液2次。当细胞融合至80%~90%后,以1∶2的比例传代至第3代时用于后续实验。一抗选择Vimentin,二抗选择荧光(Cy3)标记羊抗兔IgG荧光,免疫荧光检测细胞类型。
1.2.3纳米粒材料制备及牙周膜细胞转染效率检测 在课题组前期研究中,已成功使用2,2′-二硫二乙醇对硝基苯基碳酸酯和含有叔胺的二伯胺类化合物 1,4-双(3-氨基丙基)-哌嗪,通过缩聚反应制备出含有双硫键的阳离子型聚氨酯 [2,2′-dithiodiethanol bis (p-nitrophenyl carbonate) and 1,4-bis(3-aminopropyl) piperazine,BAP],且已证明这是一种基因传递的、安全高效的非病毒载体[11]。因此,本实验选择BAP搭载lincRNA-EPS过表达质粒形成转染复合物。提前1 d将mPDLCs置于6孔板中铺板,铺板密度为2.5×105个/孔,混匀,贴壁培养。24 h后细胞融合至60%~70%时进行转染。取100 μL的Opti-MEM培养液,加入1.5 mL无菌EP管中,然后加入1 μg相应质粒,混匀后加入BAP,再次混匀后室温静置20 min。将质粒混合物加入细胞培养物中,放入培养箱中继续培养。培养24 h后,采用流式细胞术检测转染效率。
1.2.4mPDLCs转染及LPS诱导 共设5个实验组:野生型对照组(WT组)、野生型刺激组(WT+LPS组)、敲除型对照组(KO组)、敲除型刺激组(KO+LPS组)及挽救实验组(KO+LPS+lincRNA-EPS组)。在后续实验进行的前1 d,将细胞以1×105个/孔的密度接种于含有适量完全培养液的24孔细胞培养板中。在WT+LPS组、KO+LPS组及KO+LPS+lincRNA-EPS组中加入100 ng/mL的LPS溶液,刺激6 h。KO+LPS+lincRNA-EPS组提前使用BAP搭载lincRNA-EPS过表达质粒进行转染。WT组及KO组不做处理。
1.2.5RNA提取和RT-qPCR的使用 用TRIzol试剂提取待测孔板中实验细胞的总RNA,经紫外分光光度计测量总RNA的浓度及纯度后,使用逆转录试剂盒将其逆转录后获得cDNA。根据RT-qPCR的试剂盒说明书配制相应的反应体系,设定RT-qPCR 的循环条件:①95℃,30 s;②95℃,5 s;③60℃,30 s;步骤②和③重复40个循环。测定的炎症相关基因为 cxcl10、cxcl12、cxcl14、IL-1α 及 IL-6,以 GAPDH为内参对照。各引物序列见表1。
1.2.6ELISA测定相关细胞因子 将各组待测孔板中的细胞培养液转移至离心管,在4℃下以1500r/min离心10 min,立即分装上清液并在-80℃下保存样本,待用。使用 ELISA 试剂盒(cxcl10、IL-1α、IL-6),按照说明书要求进行检测,所有通过ELISA进行细胞因子分析的实验均按照一式三份进行。操作步骤:①以捕获抗体质量浓度为1~10 μg/mL的碳酸氢盐缓冲液包被微量滴定板的孔,在4℃下过夜;②封闭后加入100 μL稀释样品;③加检测抗体后室温下孵育2 h;④添加偶联二抗置室温下孵育1~2 h;⑤加底物液,终止反应后进行检测结果的判定和分析。
1.2.7统计学分析 采用SPSS 20.0软件包对获得的数据进行统计学分析。实验重复3次,各组数据经正态分布及方差齐性检验后使用,计量资料以均数±标准差(±s)的方式表示。采用单因素方差分析比较组间差异,P<0.05表示差异有统计学意义。
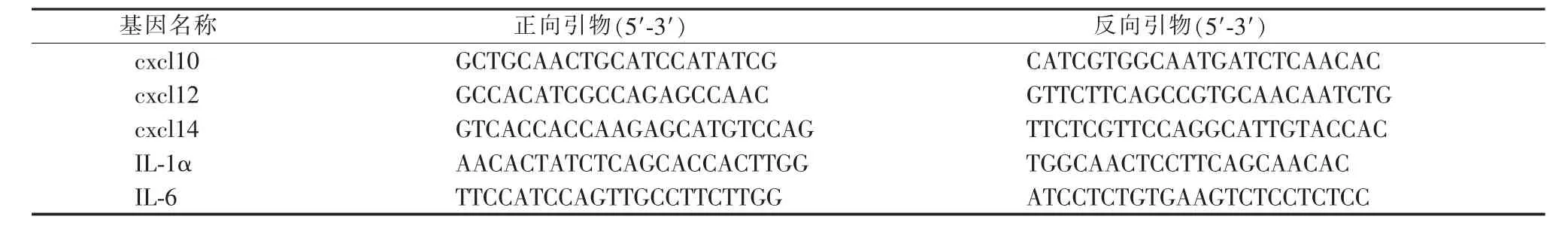
表1 炎症因子基因引物序列Table 1 Primer sequences of inflammatory cytokines
2 结果
2.1 成功构建lincRNA-EPS基因敲除小鼠模型
体外转录Cas9 mRNA和gRNA电泳结果见图1,得到gRNA目的条带与设计的gRNA一致,表明mRNA和gRNA载体构建成功。F0代小鼠PCR产物经测序比对(表2),确认获得2只目的基因缺失的阳性F0代小鼠。通过后续繁殖获得的纯合型目的基因缺失子代,经基因鉴定后可用于后续实验。
2.2 成功分离培养mPDLCs
免疫荧光结果(图2)显示,Vimentin阳性率>90%,即细胞纯度>90%。结果说明成功分离培养了mPDLCs。
2.3 转染效率测定
BAP搭载lincRNA-EPS过表达质粒成功转染mPDLCs,转染效率接近20%(图3),具有良好的转染效果,符合后续实验需求。
2.4 lincRNA-EPS敲除对牙周膜细胞炎症因子mRNA表达的影响
RT-qPCR结果如图4所示,KO组各炎症因子mRNA的表达量较低,与WT组相比,无显著性差异(P>0.05);LPS刺激 6 h后,KO+LPS组相比 KO 组,以及WT+LPS组相比WT组,炎症因子的表达均上调(P<0.05),且KO+LPS组表达水平相较于WT+LPS组更高(P<0.05);而挽救实验组相对于KO+LPS组,各个炎症因子的mRNA表达量显著下调(P<0.05)。
2.5 lincRNA-EPS敲除对牙周膜细胞炎症因子蛋白表达的影响
ELISA结果如图5所示,KO+LPS组相比WT+LPS组,cxcl10、IL-1α的蛋白表达水平更高 (P<0.05),而 IL-6 无差异(P>0.05);挽救实验组与 KO+LPS 组比较,其 cxcl10、IL-1α、IL-6 的蛋白表达水平均显著下降(P<0.05)。

图1 体外转录Cas9 mRNA和gRNA的电泳结果Figure 1 Electrophoresis of Cas9 mRNA and gRNA in vitro

表2 F0代小鼠PCR产物测序结果Table 2 Sequencing results of F0 generation mouse PCR products
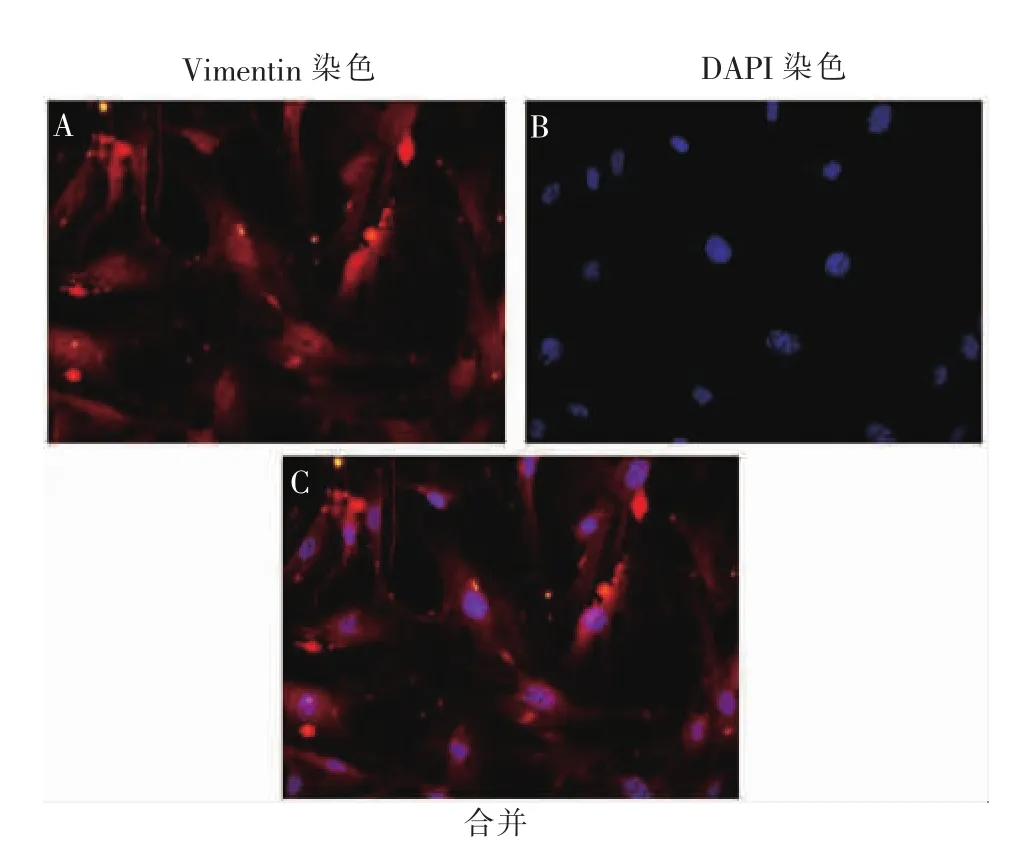
图2 mPDLCs免疫荧光鉴定(×400)Figure 2 Immunofluorescence identification of mPDLCs(×400)
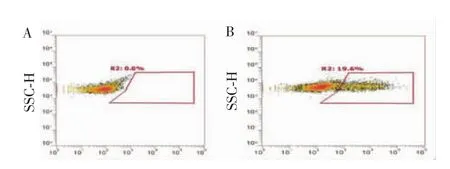
图3 流式细胞术检测BAP转染效率Figure 3 Transfection efficiency of BAP detected by flow cytometry
3 讨论
牙周炎是口腔常见的一种慢性感染性疾病,严重影响口腔健康甚至影响全身健康。LPS作为牙周炎主要致病菌(革兰氏阴性杆菌)的毒力因子,不仅能直接作用于牙周组织细胞,引起牙周组织的破坏,同时也是一种潜在的细胞激活因子,可促进牙周组织的炎症反应和骨吸收,在牙周炎的发生、发展过程中起重要作用[6,12]。TLR4作为LPS的受体,是Toll样受体家族成员之一,在引发炎症反应、促进免疫细胞成熟分化及调节免疫应答等方面起重要作用[13-14]。当TLR4成功识别LPS后,首先会引起一系列接头蛋白的招募,进一步可以向下传递级联信号,最终通过磷酸化作用,使转录因子活化并进入细胞核,从而激活包括促炎性细胞因子(IL-1、IL-6、TNF-α 等)和趋化性细胞因子(IL-8)在内的多种细胞因子的表达。可见,牙周炎的发展与LPS介导的宿主免疫反应是密切相关的。
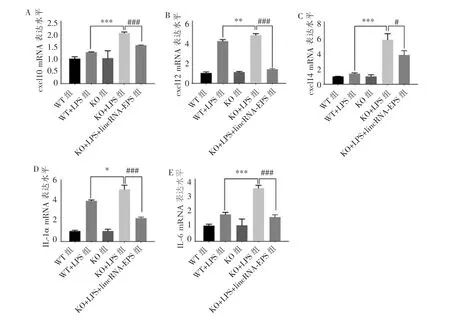
图4 各组mPDLCs炎症因子mRNA的表达水平Figure 4 mRNA expression level of inflammatory cytokines in mPDLCs
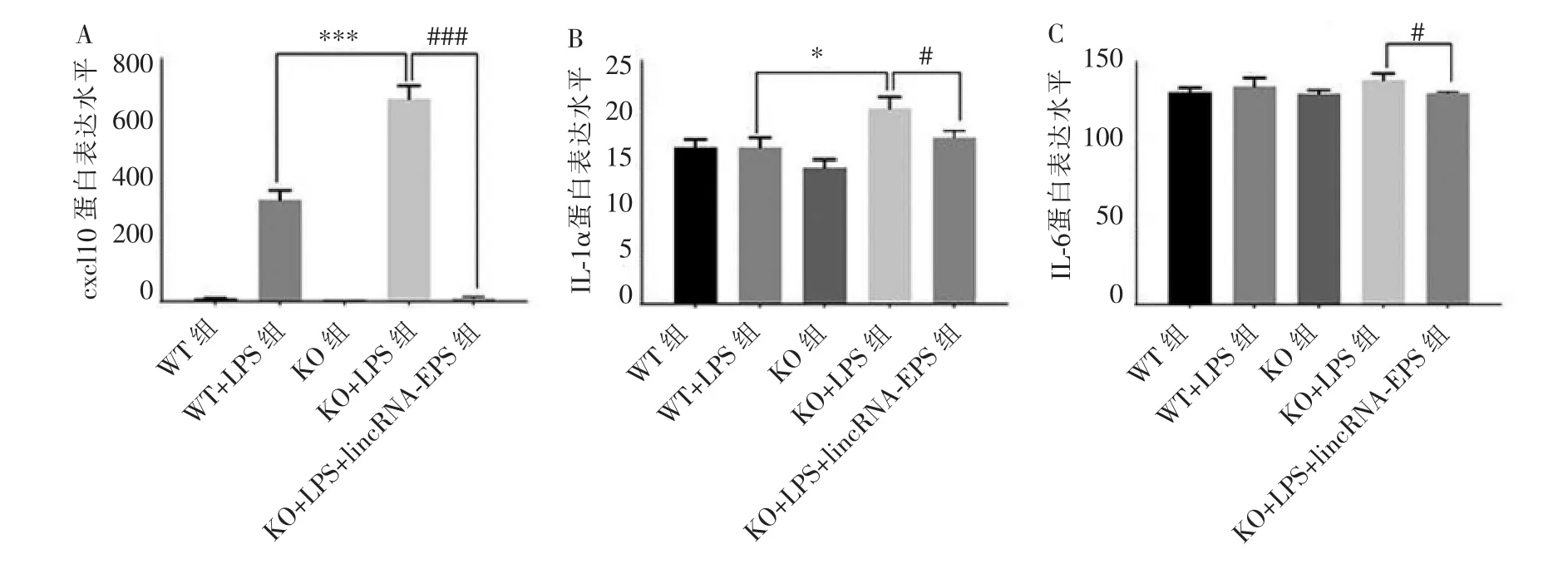
图5 各组mPDLCs炎症因子蛋白的表达水平Figure 5 Protein expression level of inflammatory cytokines in mPDLCs
LncRNAs可通过介导染色体重塑、组蛋白修饰、X染色体失活,以及基因组印迹、转录、剪接、翻译、降解和转运等途径,在表观遗传水平、转录水平和转录后水平等层面上调控基因的表达,从而影响疾病的发生和发展[15-16]。近年来的研究发现,lncRNAs不仅在炎症反应中发挥作用,而且还是调控免疫功能的关键因子[17]。而牙周炎也是免疫系统功能异常引发骨组织破坏的疾病之一。Zou等[18]通过基因芯片分析发现,相较于邻近正常组织,慢性牙周炎组织中表现出明显的lncRNAs差异性表达,提示lncRNAs可能在牙周炎的发病和发展过程中起重要作用。研究发现,lincRNA-EPS能精确调控IRGs的表达,lincRNA-EPS定位于IRGs调控区,可控制核小体定位并抑制IRGs转录,lincRNA-EPS缺乏小鼠在受到LPS刺激后,心脏、肝脏、脾脏、肾脏组织均表现出明显的炎症反应,而引入外源性lincRNAEPS后,炎症反应明显减轻[10]。另外,有研究表明,牙周组织先天性免疫系统是口腔抵抗外来侵害的一道屏障,而该过程是依赖于相关受体识别微生物或炎症信号后引起的转录变化,通过受体信号转导给下游的转录因子后诱导成千上万的免疫IRGs表达,进而引起免疫反应[19]。而由LPS诱导的牙周炎,是否也受到lincRNA-EPS的调控,目前尚未见文献报道。
本实验从功能缺失和功能获得2个方向研究lincRNA-EPS在LPS诱导的mPDLCs中的调控作用。CRISPR/Cas9是一种高效、快速、便捷、廉价的基因编辑工具,可靶向敲除基因[20]。此技术已成熟应用于lncRNA基因敲除动物模型的构建[21]。本实验结果表明,应用CRISPR/Cas9技术能成功构建lincRNAEPS基因敲除的C57BL/6J小鼠。研究已证明,IL-6、IL-1α在慢性牙周炎的发生、发展过程中起作用[22]。且有研究证明,cxcl家族在介导LPS诱导的牙周炎中起重要作用,cxcl10在牙周炎患者中的表达显著增加,可视为一种生物标志物[23-24]。本研究中,我们应用RT-qPCR和ELISA技术检测各实验组牙周膜细胞IL-6、IL-1α、cxcl10的表达。检测结果发现,各炎症因子在WT组和KO组中表达量较低,2组数据经统计学分析,结果无显著差异。这表明,在无LPS刺激的正常状态下,lincRNA-EPS的缺失不会直接导致牙周膜细胞炎症因子基础表达量的增高。WT+LPS组和KO+LPS组与各自的对照组相比,其IL-6、IL-1α、cxcl10的表达明显增高,且差异具有统计学意义(P<0.05)。这表明LPS刺激成功诱导了mPDLCs炎症因子的表达上调。而KO+LPS组相较于WT+LPS组,各炎症因子的表达量更高,且差异具有统计学意义(P<0.05)。这表明lincRNA-EPS缺失的mPDLCs在LPS诱导下会产生更强烈的炎症因子表达,而lincRNA-EPS的正常表达则可在一定程度上抑制炎症因子的表达。目前,基因治疗被认为是牙周炎的一种替代疗法,当前的基因递送方法在转导、转染效率方面显示出可靠、一致的结果[25]。目前,已有研究将纳米颗粒载体应用于牙周膜细胞的基因递送[26-27]。在课题组的前期研究中已成功合成BAP,且证明其具有良好的转染效果。因此,本实验选择BAP作为质粒载体。流式细胞术检测结果显示,BAP搭载lincRNA-EPS过表达质粒形成的转染复合物具有较好的转染效果。本实验通过BAP搭载lincRNA-EPS过表达质粒成功转染mPDLCs,构建了挽救实验组,挽救实验组相较于KO+LPS组,各炎症因子表达量明显下降,且差异具有统计学意义(P<0.05)。这说明外源性lincRNA-EPS的引入有效抑制了炎症因子的表达,也证明以lincRNA-EPS为靶向基因的基因治疗存在一定的可行性。
综上所述,牙周膜细胞中lincRNA-EPS的缺失,可导致LPS诱导的牙周膜细胞中炎症因子的表达明显增强,而通过引入外源性lincRNA-EPS的方法则有效抑制了炎症因子的表达,表明lincRNAEPS可能在LPS诱导的牙周炎症中起到负向调控作用。本研究初步验证了lincRNA-EPS在LPS诱导性牙周炎中可能起到重要的负向调控作用,并为探究lincRNA-EPS与牙周炎之间的关系及可能的相应基因治疗,奠定了初步的实验基础。
