脂毒性在非酒精性脂肪性肝病发生发展中的作用
2021-03-03张丽慧刘鸣昊赵文霞
张丽慧, 刘鸣昊, 赵文霞
河南中医药大学第一附属医院 脾胃肝胆病科, 郑州 450000
非酒精性脂肪性肝病(NAFLD)是目前最常见的慢性代谢性肝病之一,已成为世界性的公共卫生问题,其疾病谱包括非酒精性肝脂肪变、非酒精性脂肪性肝炎(NASH)、肝硬化和肝细胞癌[1-2]。我国NAFLD整体患病率为1%~30%(中位数10%)[3]。高达30%的非酒精性肝脂肪变患者会进展为NASH,出现肝细胞损伤、炎症和纤维化的病理表现[4]。脂肪变性本身不会对NAFLD预后产生负面影响[5],炎症、纤维化决定了NAFLD的长期预后[6]。越来越多的研究显示,脂毒性是促进NAFLD进展的重要机制。本文重点介绍了脂毒性在NAFLD发生发展中的作用,旨在为NAFLD的防治提供新的切入点。
1 脂毒性概述
脂毒性是指脂质在肝脏、心脏、肾脏、肌肉、胰腺等非脂肪组织中蓄积,并对这些器官或系统产生有害作用[7]。自1994年Roger Unger首次描述脂毒性以来,研究者对脂毒性在NAFLD中的危害性认识逐渐深入。肝脏脂质的来源包括饮食中残留的乳糜微粒,甘油三酯(TG)代谢释放的游离脂肪酸(free fatty acid,FFA)和相关新生脂肪等。TG是NAFLD中最多的脂质类型,而TG本身并没有脂毒性[8],具有脂毒性的特异性脂质是除TG以外的脂质所产生的,如FFA、甘油二酯、神经酰胺、溶血磷脂酰胆碱和游离胆固醇等[9]。特异性脂质引起的脂肪变性是导致肝脏脂毒性的病理基础。特异性脂质在肝组织沉积后,可引起细胞应激、功能障碍,最终导致肝细胞死亡[10]。一项对NASH小鼠模型的研究[11]表明,抑制TG合成尽管降低了肝脂肪变性的程度,却增加了肝脏FFA的沉积及肝损伤和纤维化的严重程度。肝脏脂质代谢紊乱可引起TG过度脂解,FFA释放并进入血液循环。FFA酯化为TG,是抵抗肝脏脂毒性损害的一种保护性机制[12],当大量的FFA酯化为TG受阻,超过肝脏的自我修复能力时,脂毒性主要来源的FFA沉积在肝脏,即可促进NASH和相关肝硬化的发生。
2 脂毒性驱动NAFLD发生发展的机制
目前已有研究[13]显示,脂毒性可触发肝脏内质网应激、细胞死亡、炎症等病理反应,引起脂毒性肝损伤,导致NAFLD进一步出现炎症、纤维化。
2.1 脂毒性与肝脏内质网应激 肝脏内质网可以维持正常代谢功能,许多致病因素会扰乱肝细胞内质网的稳态,导致肝脏脂质代谢失调和各种肝病的发生[14]。内质网应激在NAFLD中会变为慢性,诱导炎症反应和细胞死亡而促进其向NASH甚至肝硬化、肝细胞癌发展。研究[15]表明肝脏内质网应激与NAFLD发病密切相关。FFA在肝脏大量聚集,产生脂毒性,并通过上调蛋白激酶C-δ[16],破坏内质网稳态机制,导致内质网中错误或未折叠的蛋白质积聚,并通过3种内质网跨膜传感器启动的“未折叠蛋白反应”(unfolded-protein response,UPR)触发内质网应激[17]。肝脏持续或过度的内质网应激使肝细胞自身修复能力不足,抵抗外界刺激时,激活内质网超负荷反应,继而引发脂肪堆积、炎症、胰岛素抵抗和细胞凋亡等反应,促进了NAFLD的发生发展。3种内质网应激传感器触发内质网应激,加重肝脏脂肪变的机制如下。
内质网应激传感器肌醇依赖酶1α(inositol-requiring enzyme 1α,IRE1α)的激活可导致c-Jun氨基末端激酶(c-Jun N-terminal kinase,JNK)磷酸化和X-box结合蛋白1(X-box binding protein 1,XBP-1)两条主要途径的激活,其中XBP-1是对UPR适应性反应的关键调节因子[18]。IRE1α-XBP-1信号轴通过调节极低密度脂蛋白的分泌和脂肪的生成在肝脏脂质代谢中起着关键作用[19]。特异性肝细胞IRE1α缺失的小鼠经衣霉素(激活内质网应激的诱导剂)攻击后,由于脂质合成调节剂(C/EBPβ、C/EBPβγ、PPARγ)的抑制和极低密度脂蛋白的分泌停止,导致肝脏脂肪变性增加[20]。XBP-1在NAFLD形成过程中具有明显的激活巨噬细胞作用[21],还可通过启动子结合直接激活固醇调节元件结合蛋白-1c等生脂基因促进肝细胞脂肪形成[22]。
内质网应激传感器蛋白激酶R样内质网调节激酶(protein kinase r-like ER kinase,PERK)是一种跨膜蛋白,其主要底物是真核细胞翻译起始因子eIF2α[23]。PEPK-eIF2α信号轴通过调节活化转录因子(activating transcription factor,ATF)4的翻译,促进新生脂肪的生成并加重肝脂肪变性[24-25]。有报道[26]显示ATF4缺乏症可减轻高碳水化合物饮食小鼠肝脂肪变性,且ATF4基因缺失的小鼠可免受年龄相关和饮食诱导的肥胖及脂肪变性的影响。
内质网应激传感器ATF6的信号轴可能对脂肪变性有保护作用。全身ATF6α基因敲除小鼠表现出饮食诱导的肝脂肪变性和葡萄糖耐量的加重,并且ATF6α基因敲除小鼠经内质网应激诱导剂衣霉素攻击后,出现持续转录因子CAAT/增强子结合同源蛋白(transcription factor CAAT/enhancer-binding homologous protein,CHOP)表达、C/EBPα抑制和肝脂肪变性[27]。研究[28]显示,在肝细胞中,ATF6可与PPARα相互作用,增强肝脂肪酸氧化的转录活性,进而减轻肝脂肪变性。
2.2 脂毒性与肝细胞死亡 脂毒性导致细胞功能障碍,最终导致细胞死亡。细胞死亡的形式有3种:凋亡、程序性坏死和焦亡。凋亡是NAFLD中最常见、最具特征性的细胞死亡形式,过度的肝细胞凋亡是NAFLD的病理学标志[29],肝细胞凋亡的程度与炎症活动程度和纤维化分期相关[30]。NAFLD的细胞凋亡有3种途径,分别是死亡受体途径、线粒体损伤途径和内质网应激途径。
肿瘤坏死因子相关凋亡诱导配体受体2(TNF-related apoptosis-inducing ligand receptor 2,TRAIL-R2)的死亡受体信号转导已成为脂毒性诱导肝细胞死亡的关键机制。研究[31]显示:具有脂毒性的饱和FFA棕榈酸酯处理肝细胞会导致质膜结构域重组,从而使TRAIL-R2聚集,并激活Caspase-8。活化的Caspase-8不足以引起蛋白水解酶级联反应时,会将Bid(一种仅含BH3结构域的Bcl-2家族蛋白)裂解成活性片段tBid[29]。tBid转移至线粒体,线粒体嵴发生重组,通透性改变,促使细胞色素C等促凋亡因子释放[32],导致肝细胞死亡。肝脂毒性可通过触发持续或过度的内质网应激,诱导CHOP的表达,进而介导TRAIL-R2的上调,通过TRAIL-R2死亡受体信号转导途径促使肝细胞死亡。此外,CHOP又可通过凋亡相关基因p53上调凋亡调节因子(p53 up-regulated modulator of apoptosis,PUMA)[33],作用于线粒体,激活细胞死亡的线粒体途径,导致肝细胞死亡。脂毒性触发内质网应激后,还可以激活细胞内应激激活激酶JNK,上调促凋亡蛋白B细胞淋巴瘤2样蛋白11(B-cell lymphoma 2-like protein 11,Bim),也通过线粒体凋亡途径促进肝细胞死亡[34]。脂毒性诱导肝细胞凋亡,上游主要由死亡受体和内质网应激途径介导,下游主要由线粒体损伤途径介导。在NAFLD发生发展过程中,细胞凋亡不仅诱导了肝脏的炎性反应,更促进了肝纤维化的形成。肝细胞凋亡后,肝星状细胞(HSC)与Kupffer细胞吞噬凋亡小体后活化,释放大量炎性细胞因子,作用于下游的核因子-κB和JNK激酶信号转导通路,形成炎性瀑布反应,活化中性粒细胞引起肝脏炎性反应。肝细胞凋亡产生的DNA是HSC活化和分化的重要介质,活化的HSC还可以分泌基质金属蛋白酶,进一步促进肝纤维化的形成[35]。
2.3 脂毒性与肝脏炎症 肝脏炎症是NAFLD的一个重要组织学标志。NAFLD的炎症发生在没有病原体或外部抗原的情况下,称为无菌炎症[36]。这种无菌炎症是脂质诱导的肝细胞应激、损伤和细胞死亡的结果。脂毒性诱发的NAFLD炎症,一种是通过诱导肝细胞死亡所致的先天免疫细胞介导的炎症反应[37];另一种是引起亚致死性肝损伤所致的胞外囊泡释放触发的异常促炎信号级联反应[13]。
脂毒性诱导的先天免疫细胞介导的NAFLD炎症特点是Kupffer细胞、单核细胞来源的巨噬细胞显著聚集,中性粒细胞和自然杀伤细胞数量增加[38],树突状细胞也参与NAFLD的炎症过程。Kupffer细胞聚集与NAFLD组织学炎症活动的严重程度呈正相关。Kupffer细胞中NOD样受体家族3炎症小体的激活诱导IL-1β的分泌,促进NASH的进展[39]。Kupffer细胞的衍生因子促进单核细胞来源的巨噬细胞大量渗透到肝脏,与Kupffer细胞一起促进NAFLD的进展[40]。在NASH小鼠模型中,经处理使单核细胞来源的巨噬细胞募集减少时,肝脏炎症和纤维化得到改善[41]。中性粒细胞浸润是NAFLD进展的重要机制之一,其释放由核酸和抗菌剂组成的中性粒细胞胞外杀菌网络,捕获病原体并限制感染[42]。在小鼠中阻断中性粒细胞胞外杀菌网络的形成可以阻断NASH小鼠向肝细胞癌的转化[43]。中性粒细胞加重NAFLD的另一个机制是通过人的中性粒细胞肽在炎症过程中诱导细胞因子和趋化因子的释放,诱导HSC的增殖而加重肝纤维化[44]。自然杀伤细胞可以通过肿瘤坏死因子样凋亡诱导配体的产生,加重脂肪变性肝脏的炎症状态,并通过这一机制促进非酒精性肝脂肪变向NASH进展[45]。树突状细胞如何参与NAFLD的炎症过程尚不明确,有限的研究显示树突状细胞的耗竭导致了NAFLD肝纤维化和炎症的改善或加重,这种矛盾结果的出现可能是由于肝树突状细胞的异质性和用于实验操作树突状细胞方法的低特异性所致[12]。
胞外囊泡是脂毒性诱导细胞间信号转导的介质[46]。NASH小鼠模型和患者中,外周循环的胞外囊泡显著增加[47]。将从高脂饲料喂养的小鼠血清中分离的胞外囊泡转移到普通饲料喂养的小鼠体内,可导致普通小鼠肝脏IL-6、TNFα等促炎因子和血清ALT、AST水平升高[48-49]。不同物种的肝细胞经具有脂毒性的脂质如棕榈酸酯处理后,可释放出更多的胞外囊泡[46],这些胞外囊泡以C-X-C基序趋化因子配体10依赖的方式诱导巨噬细胞活化,通过TRAIL依赖的机制激活巨噬细胞[50],引发NAFLD肝损伤和炎症反应。最近的一份报告[51]表明,棕榈酸诱导的胞外囊泡释放是由UPR传感器-IRE1α所介导,这些胞外囊泡富含C16∶0神经酰胺,并通过神经酰胺衍生的鞘氨醇-1-磷酸信号通路促进巨噬细胞的活化,进一步强调了胞外囊泡上的脂质物质在NAFLD炎症损伤中的作用。
脂毒性触发内质网应激、细胞死亡、炎症,驱动NAFLD发生发展的机制详见图1。
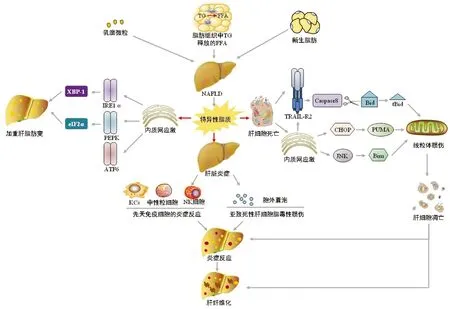
图1 脂毒性在NAFLD中的作用机制
3 总结与展望
脂毒性是NAFLD多因素发展机制中的关键因素之一,研究表明脂毒性可通过触发内质网应激加重肝脂肪变性,触发肝细胞死亡诱发肝细胞凋亡,触发炎症反应并向肝纤维化转化,促进NASH及相关肝纤维化、肝硬化的发生。脂毒性触发3种病理反应的机制大部分是在动物及细胞实验中完成的,其在人体中的作用机制如何,所触发的3种病理反应是单独发生还是同时发生?脂毒性驱动NAFLD向炎症、纤维化进展后,是否也参与NASH向肝细胞癌的转化?炎癌转化的机制如何?尚需进一步研究。深入探索脂毒性驱动NAFLD发生发展的作用机制,将为防治NAFLD提供新的切入点,为新药研制提供新的思路。
作者贡献声明:张丽慧负责查询文献,撰写论文;刘鸣昊参与修改论文;赵文霞负责指导撰写论文,修改论文并最后定稿。
