超高效液相色谱-紫外检测法测定婴幼儿配方乳粉中的核苷酸含量
2021-03-01王象欣魏雪冬李文斌鄂来明姜毓君
王象欣,单 艺,魏雪冬,李文斌,鄂来明,姜毓君
(1.东北农业大学食品学院,乳品科学教育部重点实验室,黑龙江 哈尔滨 150030;2.东北农业大学 黑龙江省绿色食品科学研究院,黑龙江 哈尔滨 150028;3.国家乳制品质量监督检验中心,黑龙江 哈尔滨 150028;4.天津迪科马实验设备有限公司,天津 300402)
核苷酸是由含氮的碱基、戊糖、磷酸3 种分子连接而成,人乳中游离核苷酸的含量范围在5~8 mg/100 mL[1],目前核苷酸已被确定为条件型必需营养素。核苷酸对降低感染性腹泻、改善肠道微生物菌群、调节免疫功能和脂质代谢有积极作用[2-3]。与未添加核苷酸的婴幼儿配方奶粉比较,添加核苷酸的婴幼儿配方乳粉被认为对健康婴幼儿胃肠道的生长和成熟有益。婴幼儿食用添加核苷酸的婴幼儿配方乳粉,其肠道中普雷沃菌属与双歧杆菌定植比例接近母乳喂养的婴幼儿水平,明显低于食用未添加核苷酸的婴幼儿配方乳粉的婴幼儿[4-5]。GB 14880ü 2012《食品营养强化剂使用标准》[6]中规定了婴幼儿配方乳粉中允许添加的核苷酸种类有5’-胞嘧啶核苷酸(cytidine 5’-monophosphate,CMP)、5’-尿嘧啶核苷酸(uridine 5’-monophosphate,UMP)、5’-腺嘌呤核苷酸(adenosine 5’-monophosphate,AMP)、5’-次黄嘌呤核苷酸(inosine 5’-monophosphate,IMP)、5’-鸟嘌呤核苷酸(guanosine 5’-monophosphate,GMP),允许添加的使用量为12~58 mg/100 g(以核苷酸总量计)。欧盟关于婴幼儿配方乳粉的法规Commission Directive 2006/141/EC规定了婴儿和较大婴儿及幼儿配方食品中添加核苷酸的上限为5 mg/100 kcal(约23.9 mg/100 g),并对5 种成分的添加量进行了限定。
目前,婴幼儿配方乳粉中核苷酸的检测官方方法主要有GB 5413.40ü 2016《婴幼儿食品和乳品中核苷酸的测定》[7],其他文献报道的研究方法主要有反相液相色谱联合紫外检测器[8-9]或质谱检测器[10],前处理使用沉淀蛋白结合不同的固相萃取方式。经验证,离子对试剂结合C18极性色谱柱分离,使核苷酸对流动相pH值敏感,会引起杂质对UMP或GMP的干扰;HLB固相萃取方法[9]杂质去除能力弱,阴离子交换固相萃取[11]在不使用内标的情况下回收率偏低;液相色谱-质谱联用检测方法由于不锈钢毛细管喷雾钢针对核苷酸磷酸基团的捕集吸附,导致灵敏度下降、重现性差[12]。亲水作用液相色谱(hydrophilic interaction liquid chromatography,HILIC)法适合对极性化合物的分离[13-14],但是选择合适的固定相和样品前处理方式对分析方法的开发至关重要。
本研究采用具有磺酸甜菜碱两性离子官能团固定相的HILIC方法对婴幼儿配方乳粉中5 种核苷酸进行定量分析,开发针对核苷酸这种两性化合物的二维固相萃取方式,并且完善前处理方法,旨在为乳制品中核苷酸的定量分析提供重要的参考依据。同时评估婴幼儿配方乳粉在不同加工工艺过程中的损耗以及货架期的损耗,对婴幼儿配方乳粉的研发具有指导作用。
1 材料与方法
1.1 材料与试剂
婴幼儿配方牛乳粉、婴幼儿配方羊乳粉、特殊医学用途婴儿乳蛋白部分水解配方食品、特殊医学用途婴儿氨基酸配方食品均为市售;不同工艺婴幼儿配方乳粉为委托不同工厂生产。
磷酸二氢钠溶液、磷酸二氢钾溶液、正磷酸、甲酸、氨水、核苷酸标准品(CMP、UMP、AMP、IMP、GMP) 美国Sigma-Aldrich公司;乙腈、甲醇(均为色谱纯) 美国Fisher Chemical公司;乙酸(分析纯) 天津科密欧化学试剂有限公司;去离子水(电阻率18.2 MΩ·cm)由美国Milipore公司超纯水仪制备。
双层填料(弱阴离子交换/强阳离子交换)固相萃取柱天津迪科马实验设备有限公司;0.22 μm PTFE针头过滤头(13 mmh 0.22 μm) 美国CNW公司。
1.2 仪器与设备
H-CLASS超高效液相色谱(ultra-high performance liquid chromatography,UPLC)仪、2695高效液相色谱仪(配有紫外检测器) 美国Waters公司;UV-2600紫外分光光度计 日本岛津公司 ;PB-10/C酸度计德国赛多利斯公司;QGC-12T干热式氮吹仪 上海泉岛科贸有限公司;V18R多功能台式高速冷冻离心机 澳大利亚Dynamica公司;BS210S型分析天平 德国赛多利斯公司。
1.3 方法
1.3.1 色谱条件
Syncronis HILIC色谱柱(2.1 mmh 100 mm,1.7 μm,100 Å);流动相:A为0.001%甲酸-乙腈,B为10 mmol/L磷酸二氢钠,pH 5;流速0.4 mL/min;等度洗脱:A-B(80∶20,V/V);柱温35 ℃;进样量2 μL;检测器波长254 nm。
1.3.2 核苷酸标准溶液的配制与浓度校正
采用紫外分光光度法对标准溶液浓度进行校正[11],核苷酸以酸型计,结构式见表1。分别准确称取50 mg核苷酸标准品于50 mL容量瓶中。加入40 mL去离子水,溶解后,定容至刻度。分别吸取各标准储备液1.0 mL于50 mL容量瓶中,用0.25 mol/L磷酸二氢钾缓冲液(pH 3.5,用0.1%磷酸调节)定容至刻度,按照表1测定其最大波长下该溶液的吸光度。
分别准确称取UMP 10 mg、AMP 10 mg、IMP 10 mg、GMP 10 mg、CMP 15 mg于5 个10 mL容量瓶中。加入5 mL去离子水,溶解后,定容到刻度。每种核苷酸分别吸取0.1、0.2、0.4、0.6、0.8、1.0 mL于6 个50 mL容量瓶中,加入35 mL乙腈,然后用水定容至刻度,配制成系列标准溶液。
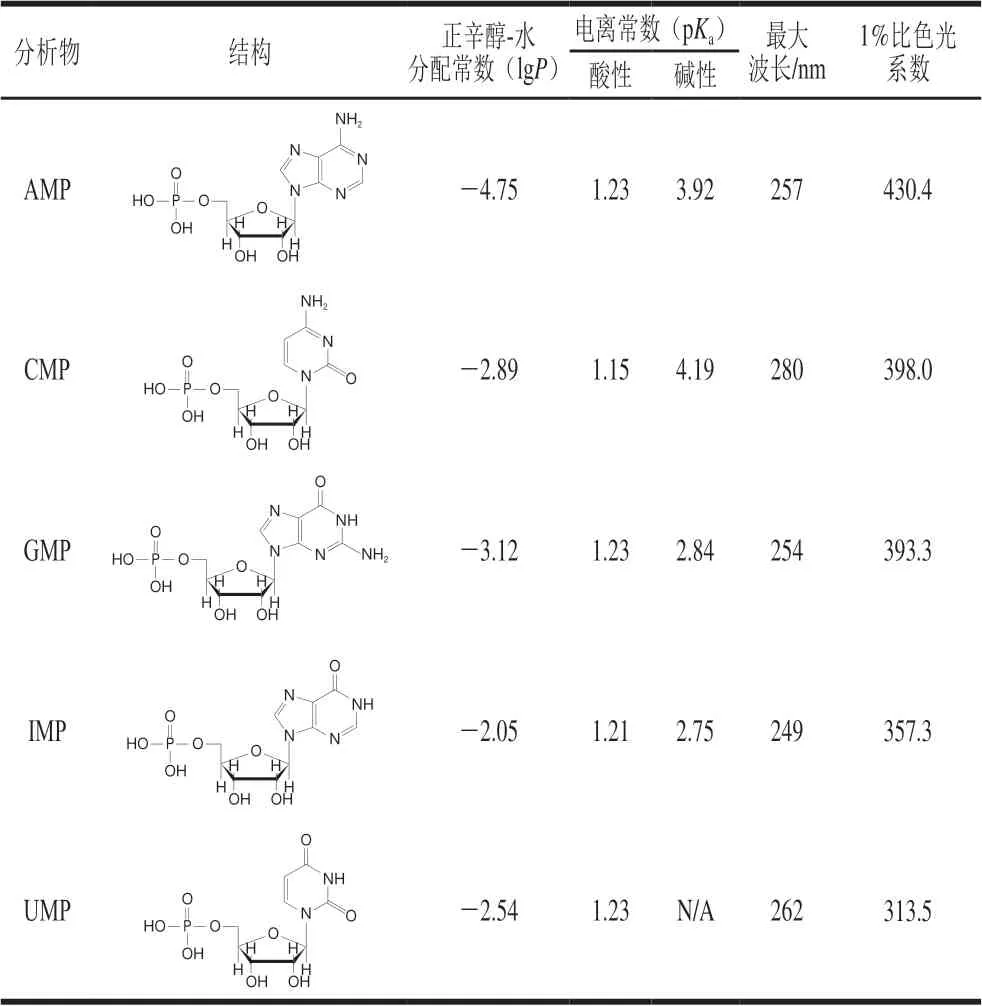
表1 5 种核苷酸的理化性质Table 1 Physicochemical properties of five nucleotides
1.3.3 样品前处理
称取混合均匀的固体试样1 g(精确至0.1 mg),于50 mL离心管中,加入10 mL超纯水(50~60 ℃)充分溶解试样,冷却至室温。用10%乙酸溶液调pH值至4.1f 0.05,转移至25 mL容量瓶中,用超纯水定容至25 mL,混匀,过滤待净化。
采用双层填料(弱阴离子交换/强阳离子交换)固相萃取柱,依次向柱中加入3 mL甲醇和3 mL水进行活化,弃去流出液;准确吸取2.5 mL待净化液加入柱中,弃去流出液;加入5 mL甲醇进行淋洗,控制流速1 滴/s;加入10 mL 30%氨水-甲醇溶液洗脱,收集流出液,控制流速1 滴/s;将洗脱液在50 ℃氮气保护下吹干,准确加入1 mL 30%乙腈溶液,超声溶解,0.22 μm PTFE微膜过滤供UPLC分析。
1.3.4 计算方法
1.3.4.1 5’-核苷酸标准品纯度计算
5’-核苷酸标准品纯度按式(1)计算:
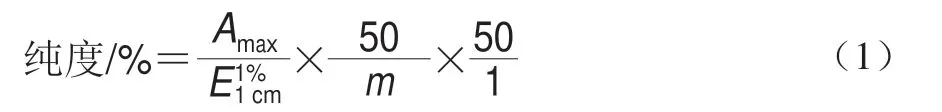
式中:Amax为在最大波长下的吸光度;为核苷酸1%比色光系数;m为标准储备液中核苷酸的质量/g;50为标准储备液的定容体积/mL;50为纯度校正溶液的定容体积/mL;1为加入到纯度校正溶液中标准储备液的体积/mL。
1.3.4.2 样品中5’-核苷酸含量计算
根据测定原理建立数学模型,见式(2):

式中:Xi为试样中核苷酸各组分含量/(mg/100 g);Ci为标准曲线查得试样溶液中各核苷酸各组分的质量浓度/(µg/mL);Vi为试样溶液体积/mL;n为稀释倍数;m为试样质量/g。
试样中游离核苷酸的总量按式(3)计算:
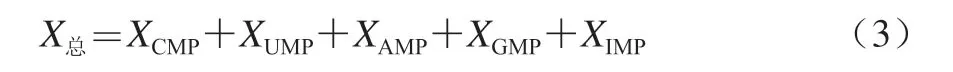
式中:X总为试样中游离核苷酸总量/(mg/100 g);XCMP、XUMP、XAMP、XGMP、XIMP分别为按式(2)计算出的对应游离核苷酸含量/(mg/100 g)。
1.4 数据统计及图表绘制
采用ChemDraw软件绘制原理结构图;理化参数(5 种核苷酸的正辛醇-水分配常数(lgP)、电离常数(pKa)、pH值依赖的正辛醇-水分配系数(lgD)和不同pH值下5 种核苷酸的预测离子化状态百分比)根据ChemAxon软件模拟计算得出;采用Excel进行数据的分析、统计及趋势图的绘制。
2 结果与分析
2.1 色谱柱的选择
2.1.1 不同类型色谱柱上核苷酸的保留情况
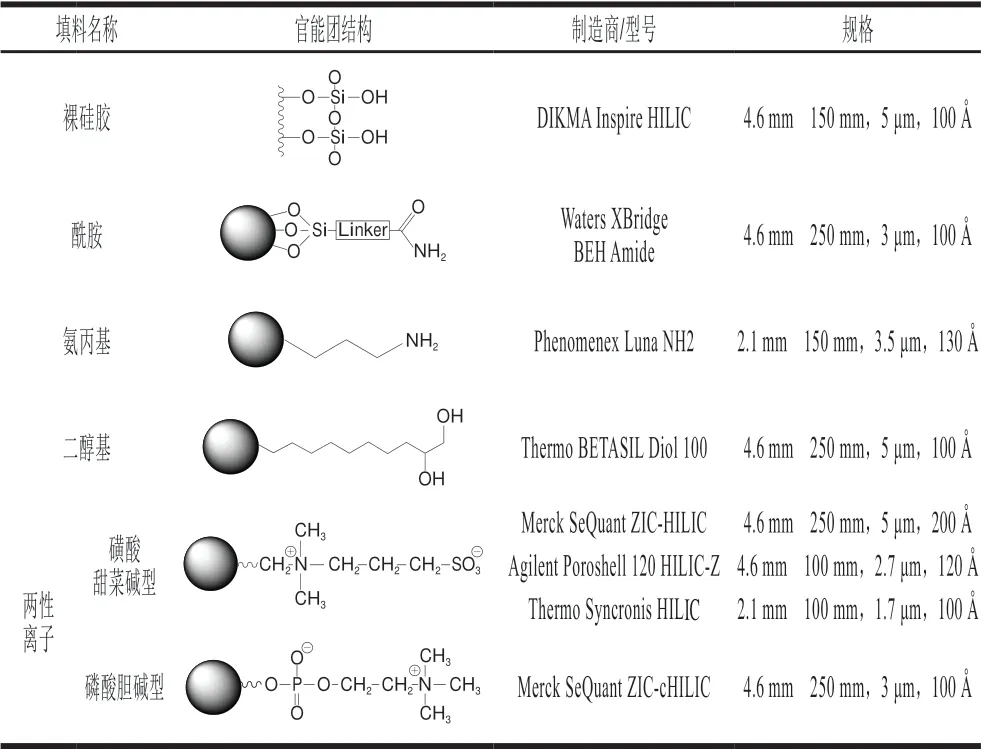
表2 不同类型HILIC色谱柱的官能团结构Table 2 Structures of functional groups of different types of HILIC columns
HILIC法为反相离子对液相方法提供了可行的替代方法,是分离和保留极性或可电离化合物(即lgP低于0的化合物,表1)的最成功方法之一。本实验测试来自6 个制造商的8 种类型的HILIC色谱柱,固定相涵盖了多种HILIC常用的键合相。如表2所示,氨丙基和两性离子色谱柱具有带电官能团,而裸硅胶、酰胺基和二醇基色谱柱在本研究使用的色谱条件下为中性。分别从保留、选择性、峰形和分离度等方面对核苷酸在不同HILIC色谱柱上的分离行为进行比较。结果表明,在核苷酸分离方面,具有不同官能团结构的HILIC色谱柱之间存在显著差异,对固定相和流动相之间的相互作用也进行了阐述。
从理论上讲,具有酰胺基和氨丙基官能团的固定相应当最适合对酸性分析物进行分离,酰胺基和氨丙基分别可与核苷酸上带负电荷的磷酸基团产生氢键和静电相互作用,从而使其在色谱柱上得到良好保留;裸硅胶和二醇基可以通过氢键作用力与带负电荷的分析物相互作用[15-16]。但从本实验结果分析发现,具有裸硅胶和二醇基官能团的色谱柱对核苷酸并没有明显保留;氨丙基色谱柱上未观察到核苷酸的洗脱,推测核苷酸可能仍保留在色谱柱上,酸性条件使氨丙基带正电,而且使固定相表面极性基团质子化,从而与核苷酸磷酸基团之间产生较强的相互作用,阻止了核苷酸的洗脱。据文献报道,氨丙基色谱柱上保留了核苷酸[17],但洗脱条件(pH 9.45)可能加速硅胶基质的降解,长时间使用可能会加快色谱柱的损坏;酰胺基色谱柱和磷酸胆碱型两性离子键合的色谱柱对核苷酸有保留,但未将5 种核苷酸完全分离,样品分离度差。
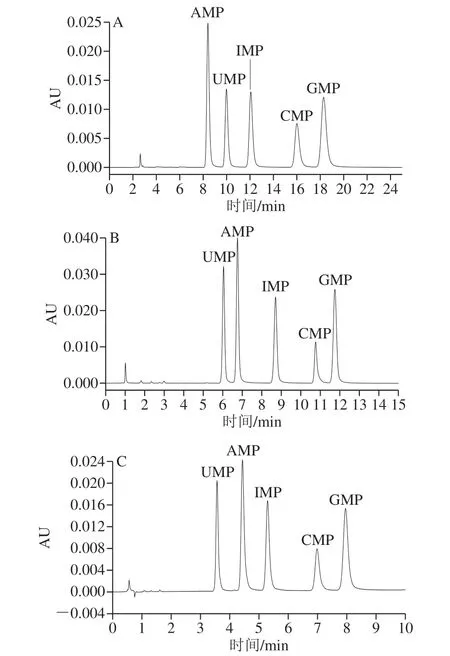
图1 5 种核苷酸在不同色谱柱上的分离情况Fig.1 Separation profiles of five nucleotides on different columns
实验过程中观察到5 种核苷酸在全多孔(Merck SeQuant ZIC-HILIC)与表面多孔(Agilent Poroshell 120 HILIC-Z)和超高压色谱柱(Thermo Syncronis HILIC)上的出峰顺序不一致(图1),可以判断出即使相同类型的两性离子磺酸甜菜碱色谱柱,在不同生产商和不同工艺下生产的色谱柱也有差别,原因可能是连接磺酸基团和磷酸基团的短链烷基长短的差异影响了2 个基团的电荷距离。而在3 款磺酸甜菜碱型HILIC色谱柱上,5 种核苷酸分离度均大于1.5,且峰形尖锐对称(图1)。综合分离时间及响应值等因素,选用Syncronis HILIC色谱柱,其具有分辨率高、分析速度快等优势,而且样品批间测定重复性高。
2.1.2 核苷酸在磺酸甜菜碱型HILIC色谱柱上保留原理的分析
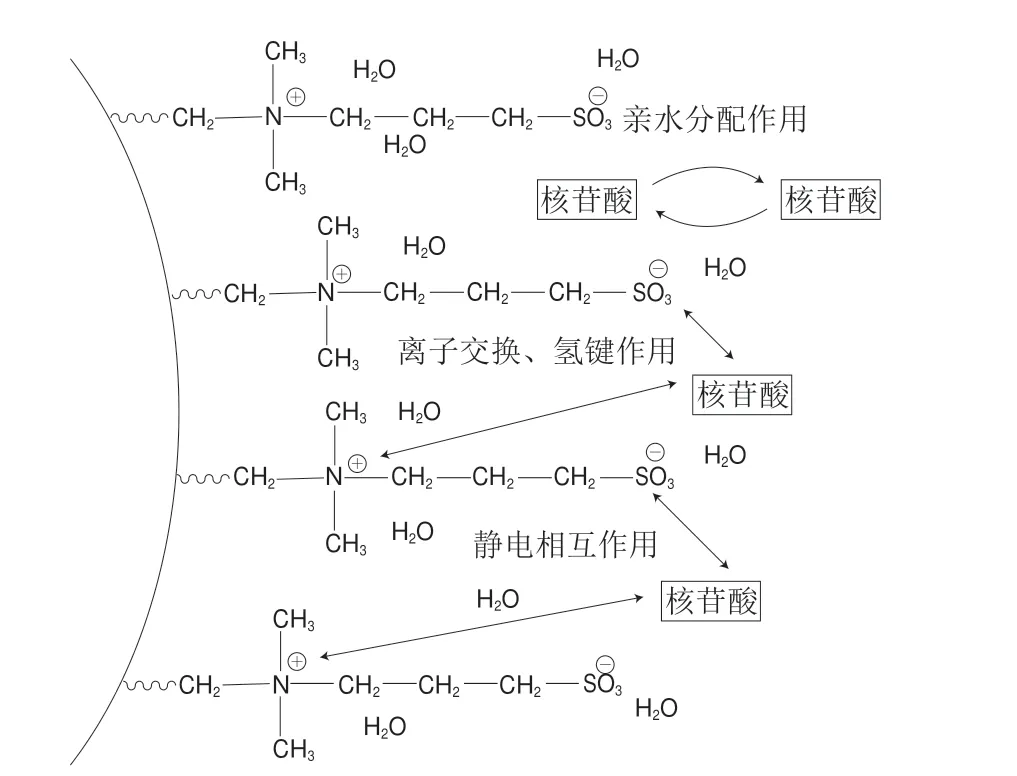
图2 5 种核苷酸在磺酸甜菜碱型两性HILIC色谱柱上的分离机理Fig.2 Separation mechanism of five nucleotides on sulfoalkylbetainebased zwitterionic HILIC column
磺酸甜菜碱型HILIC色谱柱的特点为强酸性磺酸基团(键合配体末端)和强碱性季铵基团(靠近硅胶表面)分别带有负电荷和正电荷,2 个带相反电荷的基团被一个短链烷基间隔,其物质的量比为1∶1,因此键合层上的表面净电荷非常低,受pH值的影响很小。对于两性离子固定相,离子交换和静电相互作用较典型的离子交换(氨丙基、裸硅胶、酰胺等)弱。磺酸甜菜碱键合相通过氢键强烈吸附水,大部分水层成为固定相的一部分(图2),该富水层在保留机理方面起着重要作用,使分析物在流动相和富水层之间分配;核苷酸的杂环基团(含有带正电的质子化氮原子和非质子化氮原子)和带负电的磷酸基团分别可与位于配体两端的磺酸基团和季氨基产生氢键作用和静电相互作用;固定相与核苷酸之间还存在弱离子交换作用。通过亲水分配作用、氢键、静电相互作用和弱离子交换等多种作用力的协同作用[18-22],磺酸甜菜碱键合相对核苷酸的结构差异表现出了高度的选择性,保留时间随分析物极性的增加而增加。当考虑在两性HILIC分离中的保留程度时,分析物和固定相之间的静电排斥(磺酸基团与磷酸基团)也很重要,这需要通过一定浓度的缓冲盐溶液进行调节。
2.2 流动相的选择
2.2.1 不同pH值条件下核苷酸的灵敏度和离子化状态
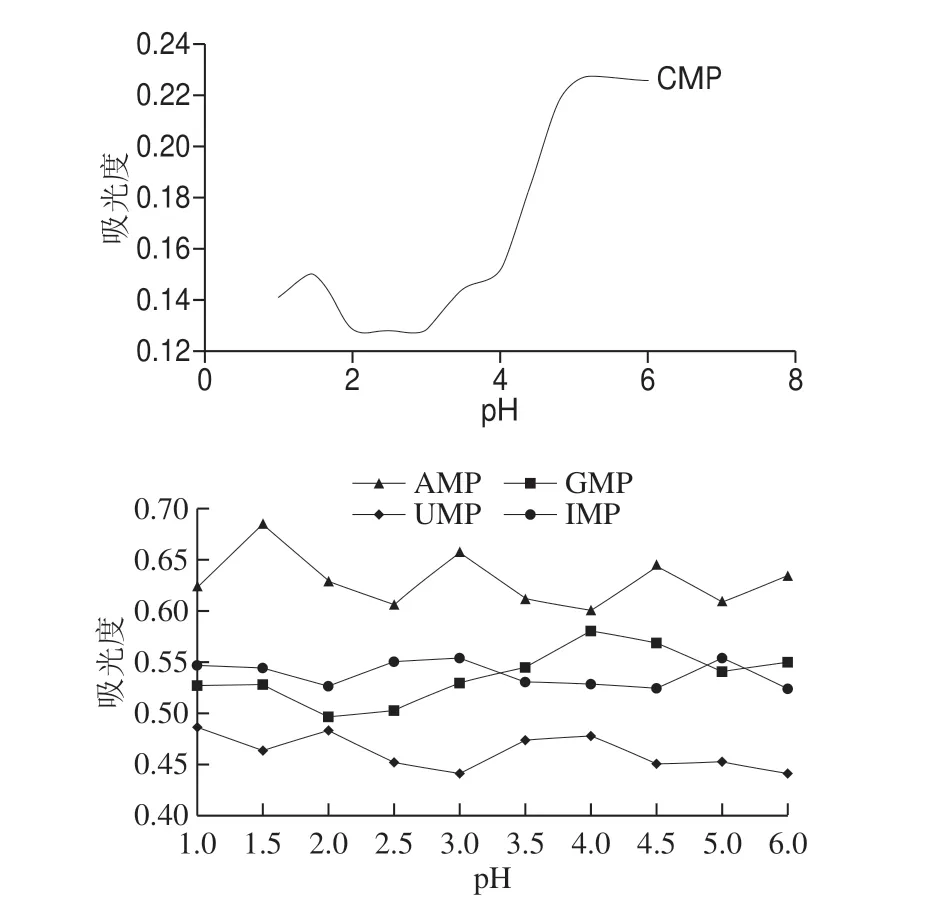
图3 5 种核苷酸在不同pH值条件下的吸光度Fig.3 Absorbance of five nucleotides at different pH values
配制pH 1~6的0.25mol/L磷酸二氢钾缓冲液,分别用不同pH值的缓冲溶液溶解同一含量的核苷酸,在波长254 nm处测其吸光度,如图3所示。可以看出5 种核苷酸在不同pH值条件下吸光度的变化,CMP的吸光度随pH值的增大而增大,CMP的吸光度在pH 1~6范围内的变异系为23.6%;而AMP、UMP、GMP、IMP的吸光度在pH 1~6范围无显著变化,其变异系数分别为4.2%、3.8%、5.0%、2.4%。通过5 种核苷酸的吸光度分析,在pH 1~6范围内,当pH>5时,5 种核苷酸的灵敏度最高。
流动相pH值的优化对于控制不同核苷酸的离子化状态至关重要,每种核苷酸在不同pH值下存在不同的离子化状态。同种核苷酸在不同离子化状态下的色谱行为不同,混合的离子化状态会导致峰展宽和保留时间的不一致。因此,找到最佳pH值,使每种核苷酸均存在一种占主导的离子化状态至关重要。使用ChemAxon软件对每个核苷酸的正辛醇-水分配系数(lgP),pH值依赖的正辛醇-水分配系数(lgD)和pKa值进行计算,如表1、3所示。从表3得出不同pH值下5 种核苷酸的lgD值均小于0,说明在此pH值范围内5 种核苷酸均适用于HILIC分析。图4为使用ChemAxon软件计算得出的不同pH值下CMP的预测离子化状态百分比,选取占比最大的A、B和C 3 个离子化状态进行说明:A离子磷酸根带一个负电荷,嘧啶杂环上的氮原子被质子化,其占比最高的pH值为3附近;B离子只有磷酸根带一个负电荷,嘧啶基团无被质子化氮原子,其占比最高的pH值约为5.5;C离子磷酸根带2 个负电荷,嘧啶基团无被质子化氮原子,其占比最高的pH值为9.5~10.5。从这些数据可以明显看出,在pH 5.5的条件下CMP以单一化离子状态存在且占比最大;在pH 3条件下CMP以两性状态存在;pH 9.5~10.5的条件下硅胶基质的溶解度增加,因此对于分析CMP,其最佳pH 5.5。基于相似的分析,每种核苷酸都有自己的最佳pH值,表3数据根据ChemAxon软件计算得出,比较5 种核苷酸后,结合上述5 种核苷酸的pH值-吸光度结论,得出满足此5 种核苷酸最佳范围且最主要以单个离子形式存在的最合适的pH值为5。

表3 不同pH值下5 种核苷酸的lgD、电荷状态和电离百分比Table 3 lgD values, charge states, and ionization percentages of five nucleotides at different pH values
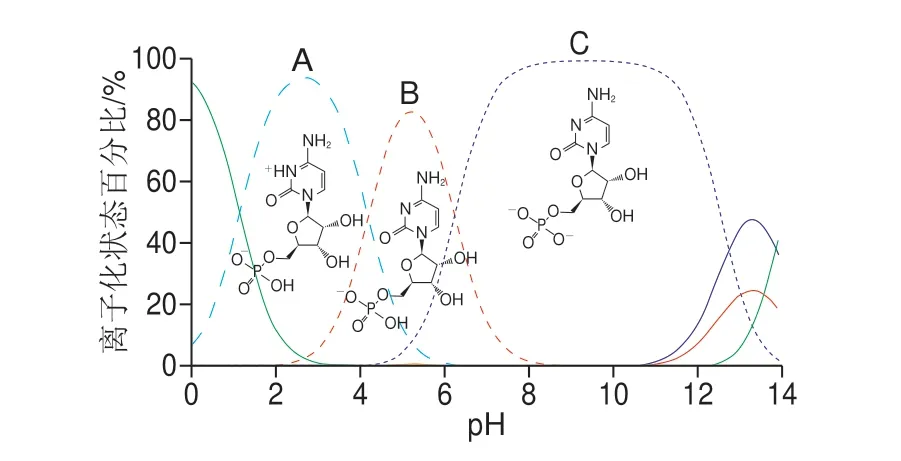
图4 不同pH值下CMP的预测离子化状态百分比Fig.4 Predicted percent ionization state of CMP at different pH values
2.2.2 缓冲盐的类型和浓度的选择
使用缓冲盐溶液,可以降低带电分析物与未修饰的带电硅醇基或固定相上带电基团之间的静电相互作用(引力和斥力)。在静电引力的作用下,缓冲盐浓度的增加会导致带电分析物在带相反电荷的固定相上的保留减少;而在静电斥力作用下,会导致带电分析物在具有相同电荷的固定相上的保留增加[17]。HILIC模式常用的缓冲盐类型有铵盐和磷酸盐,磷酸盐和乙酸铵的截止波长均小于220 nm,均适合UV检测的高效液相色谱分析。乙酸铵的缓冲范围为3.8<pH<5.8,实验过程中发现,即使乙酸铵的浓度达到100 mmol/L,5 种核苷酸也不能达到基线分离。磷酸盐具有3 个pKa值,在允许缓冲pKaf 1 pH单位的情况下,3 个缓冲范围为1.1<pH<3.1、6.2<pH<8.2和11.3<pH<13.3。使用浓度为10 mmol/L的磷酸二氢钠溶液,当其pH值在其缓冲区间2.6时,CMP响应明显降低,与上述结论相符;pH 5虽然不在磷酸盐的缓冲区间,但是5 种核苷酸分离度均大于1.5。实验过程中发现,与乙酸铵溶液相比,磷酸二氢钠溶液具有较高的电荷密度,能有效平衡分析物和磺酸甜菜碱型两性固定相之间的静电相互作用,改善了核苷酸的峰形;而且改变分析物和磺酸甜菜碱型两性固定相之间的静电相互作用所需的磷酸盐含量远低于乙酸铵溶液;此外,磷酸盐可阻断固定相基质中的痕量金属(例如铁、铜、铝等)与分析物的配位,从而改善色谱峰的形状[23]。
2.2.3 有机相的选择与优化
影响保留的主要因素是固定相类型,流动相中水溶液的比例以及水溶液中缓冲盐的类型、浓度和pH值[19,24-25]。HILIC要求使用包含质子惰性溶剂的高比例(40%~97%)有机溶剂作为流动相,以及至少3%的水或缓冲盐溶液,以配合其极性固定相,便于色谱分离。
由于甲醇可与核苷酸竞争固定相上的氢键位点,导致核苷酸的保留率和选择性降低,因此选用乙腈作为流动相中的有机相。实验过程中发现,增加乙腈的浓度,分析物在固定相上的保留时间都会延长。原因是在高比例乙腈下,流动相变得更加疏水,分析物与极性固定相之间发生了更强的相互作用,从而实现了更强的分配机制[24,26]。
在HILIC分离过程中,pH值的控制和测量较复杂。尽管准确设置水性流动相的pH值相对简单,但使用标准pH计对含有高比例有机相设置相同的pH值是不切实际的。乙腈是一种介电常数低的弱碱,大比例的乙腈会改变分析物的电离状态[26-27],也导致无法确定所需要的pH值,尤其是梯度洗脱过程中有机成分变化。实验发现,在使用0.001%甲酸-乙腈溶液代替100%乙腈溶液作为有机相时,5 种核苷酸的保留时间和分离度均有改善。
2.3 样品前处理条件的建立
2.3.1 固相萃取前样品的处理
建立固相萃取方法的过程中,样品基质的性质与目标化合物同样重要。由于HILIC模式的流动相中包含大比例有机相,使用GB 5413.40ü 2016[7]的方法进行样品前处理时,发现进样后样液遇到有机相会产生沉淀,导致柱压逐渐升高;而且,在使用同样修饰基团的表面多孔和超高压色谱柱时溶剂效应明显,CMP和GMP峰形差、峰分裂。当按照流动相初始比例稀释样液时,有沉淀产生,离心上机后测得值偏低。原因或为乙腈使蛋白质失去表面电荷而产生沉淀的同时,吸附了样液中一部分核苷酸,导致测定值偏低。根据婴幼儿配方奶粉高蛋白、高脂肪、高乳糖的性质,将样品溶解后加入一定量的甲酸溶液沉淀蛋白,分别用0.02%、0.5%、2%、4%、8%甲酸溶液进行实验,发现不同品牌的婴幼儿配方奶粉沉淀效果均不一致。后采用称量1 g样品,加入10 mL 60 ℃超纯水溶解,用10%乙酸溶液调pH值至4.1f 0.05沉淀蛋白,此种方式不论羊奶粉还是牛奶粉,沉淀效果一致,而且降低了样品的黏度,样品pH值与后续采用的固相萃取机理一致。
2.3.2 固相萃取材料的选择
乳制品的基质比较复杂,根据核苷酸的结构和pKa值(表1)及固相萃取的原理,应采用强阳离子交换(SCX)或阴离子交换固相萃取模式。但实验发现,强阳离子固相萃取模式对CMP和UMP有明显保留;弱阴离子交换对AMP、GMP、IMP有明显保留;强阴离子交换5 种成分均无法洗脱,因此通过一种模式进行固相萃取无法达到满意的效果。本研究通过以上实验结果开发出了二维固相萃取模式,即PSA(弱阴离子交换)和SCX(强阳离子交换),2 种填料在同一个固相萃取柱内添加。PSA与SCX的洗脱条件一致,均为酸性样液上柱,碱性溶液洗脱,通过PSA保留磷酸基团,SCX保留含氮基团。实验开始采用聚合物表面修饰的固相萃取填料,发现GMP回收率低,即使加大氨水-甲醇溶液中氨水的比例也不能使其全部洗脱,原因或为聚合物(苯乙烯-二乙烯基苯聚合物)材料比表面积大,表面修饰的弱阴离子交换基团多、密度高、离子交换容量为3.0 mmol/g,对GMP产生了不可逆吸附。后采用硅胶表面修饰的填料,其离子交换容量为1.8 mmol/g,提高了GMP回收率。另外采用PSA和SCX两种填料进行混合,发现回收率不稳定;SCX在上,PSA在下发现洗脱液浑浊;最终发现PSA在上,SCX在下效果最佳。
2.3.3 固相萃取方法的建立
通常采用甲醇或极性有机溶剂作为固相萃取柱的预处理溶液,水或缓冲溶液作为平衡溶液,上样后采用水或缓冲溶液和有机溶液依次进行淋洗[28]。实验发现上样后用水淋洗后,采用不同比例的甲醇溶液对固相萃取柱进行淋洗,CMP回收率均低于60%。后采用上样后直接用甲醇进行淋洗,CMP回收率有所改善。洗脱采用5%~35%的氨水-甲醇溶液依次进行实验,发现采用30%氨水(pH 10.5)作为洗脱液时5 种核苷酸回收率均大于95%。图5B与图5A相比,最后一个成分GMP出峰后仍有杂质出峰,持续20 min结束(色谱图未完全展示)。通过图5及实验数据分析,此种固相萃取方式有效的除去了杂质,保证了方法的准确性。


图5 标准品和样品色谱图Fig.5 Chromatograms of partially hydrolyzed protein infant formula samples for special medical purposes
2.4 溶剂效应的消除

图6 5 种核苷酸的溶剂效应色谱图Fig.6 Chromatograms showing solvent effect of 5 nucleotides
实验过程中发现,当使用纯水配制标准品时,溶剂效应明显(图6),原因或为样品被注入色谱柱,当样品溶剂与流动相存在差异时,一部分样品溶解进入了流动相,一部分还留在溶剂里,造成色谱保留的差异,因此使用含有一定比例乙腈的溶液配制标准品和复溶样品。由于大比例乙腈条件下核苷酸的溶解度较差,因此通过调节有机相比例和进样量2 种方式消除溶剂效应。分别使用乙腈-水和乙腈(pH 5的10 mmol/L磷酸二氢钠溶液)为60∶40、65∶35、70∶30、75∶25的溶液配制核苷酸标准溶液和复溶样品,每个标准溶液和样品分别进样1、2、3、4、5 μL。通过实验发现,使用乙腈-水比例70∶30、进样量2 μL时,无论标准品还是复溶的样品,峰形尖锐对称,有效消除了溶剂效应。
2.5 仪器条件的优化
磷酸基团是一种非常强的螯合剂,与高效液相色谱系统中的金属存在相互作用,导致色谱峰不对称,峰分裂等情况。有文献报道[29]在流动相中添加焦磷酸改善磷酸基团与金属的相互作用,但实验结果显示此方法对于核苷酸分析并不适用,因此在实验前有必要对高效液相色谱系统进行钝化。用30%磷酸溶液(取85%浓磷酸35 mL与65 mL水混匀)作清洗剂,清洗的目的是去除不锈钢管路及系统内的污垢;用6 mol/L的硝酸(取浓硝酸40 mL与60 mL水混匀)作钝化剂,钝化目的是使不锈钢管路的内表面形成光滑均匀的氧化膜,先清洗再钝化。依次用甲醇、水、30%磷酸、6 mol/L硝酸及超纯水灌注泵及进样系统,流速1 mL/min,时间1 h;待流出超纯水pH值为中性时,用纯甲醇过渡,浸润泵头及管路。
分析物和固定相相互作用的差异与HILIC模式中温度的变化有关[30-31]。将亲水性分析物从富含有机物的流动相中转移到固定相表面上的富水层是一个放热过程,而带电分析物与固定相之间的静电引力是吸热过程[25]。通常,色谱柱温度升高会导致峰变窄以及保留时间缩短[32]。本研究选取25、30、35、40、45 ℃,5 个温度对5 种核苷酸的保留因子和峰形进行比较,当柱温≤30 ℃时,5 种核苷酸的保留时间有漂移;当柱温≥35 ℃时,5 种核苷酸的保留时间稳定且峰形有所改善,灵敏度无显著差异,因此柱温选择为35 ℃。
2.6 方法验证
2.6.1 方法的线性方程、线性范围和定量限结果
分别用不同质量浓度的CMP、UMP、AMP、GMP、IMP标准工作液进样,以质量浓度为横坐标,峰面积为纵坐标,绘制标准工作曲线,通过回归计算求得回归方程和相关系数,可见在一定质量浓度范围内,质量浓度与峰面积呈正相关,相关性好,可用于外标法进行定量测定。根据GB/T 27417ü 2017《合格评定 化学分析方法确认和验证指南》[33]方法计算检出限,采用空白样品中添加目标化合物的方法,按样品前处理方法进行处理和检测,以3 倍信噪比为方法检出限,以10 倍信噪比为方法定量限,结果如表4所示。
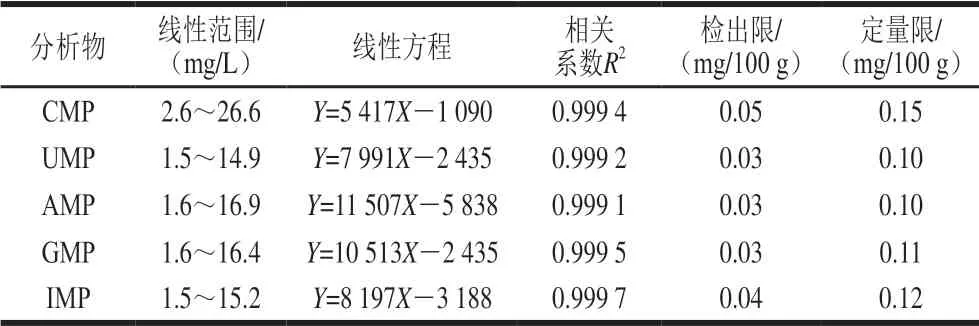
表4 5 种核苷酸的线性方程、相关系数、检出限与定量限Table 4 Linear equations, correlation coefficients, limits of detection, and limits of quantification of five nucleotides
2.6.2 方法回收率与精密度结果
根据JJF 1059.1ü 2012《测量不确定度评定与表示》[34],精密度为在规定条件下,对同一或类似被测对象重复测量所得示值或测得值间的一致程度。按照实验方法,对一定浓度含有5 种核苷酸的样品进行12 次重复性测试,计算其测试结果的标准差及相对标准偏差(变异数)。根据GB/T 32465ü 2015《化学分析方法验证确认和内部质量控制要求》[35],应使用有证标准样品核查、参加能力验证计划、与经典方法或公认方法进行比对等手段实施方法正确度研究,可结合回收率进行正确度研究,如表5所示。采用牛奶基质婴儿/成人营养奶粉有证标准物质NIST SRM 1849a作为比对样品,如表6所示,其结果均符合GB/T 27404ü 2008《实验室质量控制规范 食品理化检测》[36]中的相关要求。
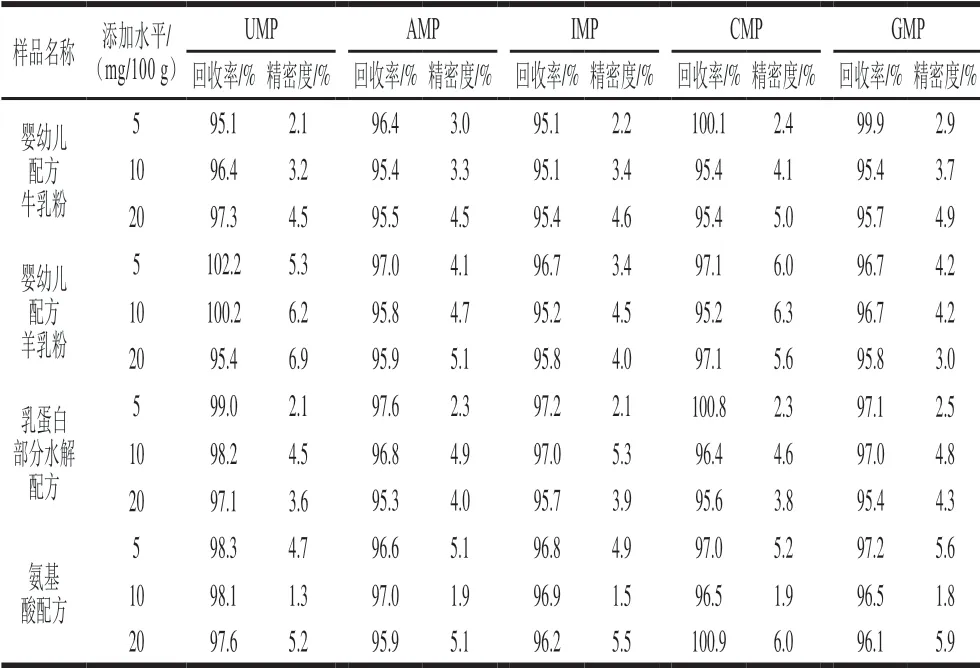
表5 不同样品中精密度与回收率结果测定结果(n=12)Table 5 Precision and recoveries for different samples (n= 12)

表6 NIST SRM 1849a测定结果(n=6)Table 6 Results of determination using NIST SRM 1849a (n= 6)
2.7 不同加工工艺过程中的损耗以及货架期损耗的评估
婴幼儿配方粉生产工艺有湿法工艺、干法工艺和干湿复合工艺3 种。在湿法工艺和干湿复合工艺下,核苷酸原料均为溶解后,经过均质、杀菌、浓缩、喷雾干燥等加工过程,应考虑核苷酸在溶解过程中的热损耗和碱性磷酸酶的分解损耗[37-38]。干法工艺下核苷酸原料直接混入基粉中,理论上无损失。通过对过保质期的产品留样测试,评估在正常储存条件下,核苷酸的损耗。使用本方法对每类样品进行测试,通过对比理论添加值与实际测试数据,发现核苷酸在婴幼儿配方乳粉的3 种生产加工过程中和货架期均无明显损耗。因此,只要确定好核苷酸原料的含水量、含盐量,同时确保核苷酸原料的添加量,即可在相应的工艺流程下生产出配方设计含量的产品。
3 结 论
本研究采用弱阴离子交换/强阳离子交换二维固相萃取法提取样品中的核苷酸,磺基甜菜碱型两性离子HILIC方法对婴幼儿配方乳粉中5 种核苷酸进行定量分析。详细阐述核苷酸在不同HILIC色谱柱上的保留情况,以及核苷酸在磺酸甜菜碱型HILIC色谱柱上保留机理,计算得出不同pH值下核苷酸的电荷状态。经过方法学验证,该方法回收率、精密度满足GB/T 27404ü 2008[36]中的相关要求。同时对婴幼儿配方乳粉在不同加工工艺过程中的损耗以及货架期的损耗进行评估,结果表明,核苷酸在婴幼儿配方乳粉的生产加工过程中和货架期均无明显损耗。本研究可为核苷酸的分离和婴幼儿配方乳粉的生产及研发提供重要的理论依据。
