TSK凝胶色谱法测定头孢克肟颗粒中的聚合物含量
2021-02-18王小明蒋婷奇谭跃浪
王小明,蒋婷奇,方 蕾,谭跃浪,陈 浩
(1.浙江巨泰药业有限公司;2.普洛药业股份有限公司:浙江 衢州 324000)
头孢克肟是一种口服的第3代头孢菌素,抗菌谱广,临床上广泛使用。头孢克肟治疗儿童细菌感染总有效率高于阿莫西林克拉维酸钾[1];用于治疗小儿急性细菌性肠炎总有效率明显高于头孢克洛,已成为儿科常用的抗菌药物[2]。但近年来,第3代头孢菌素不良反应发生率呈日趋增多,头孢类抗生素中存在的分子杂质是引发过敏反应的过敏原[3-4]。此类抗生素用于注射用时必须控制产品中的高分子聚合物的含量。
头孢克肟及其制剂聚合物检测项在各国药典中均未收载,目前《中国药典》2015版收载的头孢呋辛纳、头孢尼西钠、头孢噻肟钠等聚合物检测方法大多采用Sephadex G-10 凝胶色谱系统测定[5]。但该方法缺陷日益突出,如分析时间长,样品辅料干扰,聚合物无法完全缔合从而检出误差大等[6]。因此建立TSK gel 2500PWxl 凝胶色谱系统检测聚合物的方法。
1 实验部分
1.1 试剂与仪器
头孢克肟颗粒,生产商I,批号T41181202、T41181203、T41181204,规格50 mg;参比制剂头孢克肟细粒,生产商II,DE061;规格50 mg;头孢克肟原料,生产商III,批号AP004-1805-2048,头孢克肟的质量分数99.8%;头孢克肟对照品,生产商IV,批号130503-201706,头孢克肟计的质量分数89.2%。杂质对照品信息见表1。

表1 杂质对照品信息Tab 1 Information table of impurity reference substance
磷酸氢二钠,磷酸二氢钠,分析纯;乙腈,甲醇,色谱纯;水,超纯。
Agilent 1260 高效液相色谱仪,包括Agilent VWD、Agilent Openlab CDS 工作站;XS 105 电子天平,XPE26电子天平。
1.2 溶液的配制
样品溶液:取本品适量(约相当于头孢克肟50 mg),置25 mL 量瓶中,加甲醇5 mL,振摇溶解5 min,用水稀释至刻度,摇匀,滤过,取续滤液作为样品溶液。
对照品溶液:取头孢克肟对照品约20 mg,精密称量,置50 mL 量瓶,加甲醇5 mL,振摇溶解5 min,用水稀释至刻度,摇匀;再精密量取5 mL,置100 mL 量瓶,用水稀释至刻度,摇匀,作为对照品溶液。
系统适用性实验溶液:取样品溶液适量,于70 ℃水浴加热30 min,冷却。
1.3 色谱条件与系统适用性
以刚性、球形、亲水的多孔聚甲基丙烯酸酯树脂键合硅胶为填充剂;以磷酸盐缓冲液(0.5 mol/L磷酸氢二钠溶液、0.5 mol/L磷酸二氢钠溶液体积比61:39)与乙腈(体积比97:3)为流动相(流动相1),体积流量0.6 mL/min,检测波长254 nm,柱温30℃。精密量取系统适用性实验溶液10 μL,注入液相色谱仪,记录色谱图。
系统适用性实验溶液中杂质峰E(与头孢克肟相对保留时间约为0.89)与头孢克肟峰的分离度应不低于1.0;系统适用性实验溶液中杂质A 峰与头孢克肟相对保留时间约为0.77;与头孢克肟杂质A峰相邻的杂质E 峰与头孢克肟相对保留时间约为0.72,样品溶液中保留时间小于杂质E的聚合物杂质峰与杂质E峰的分离度应符合规定。
1.4 流动相选择
头孢克肟聚合物检测项在各国药典中均未收载,参考2015 年版《中国药典》收载的头孢地嗪钠、头孢米诺钠聚合物的检测方法,以刚性、球形、亲水的多孔聚甲基丙烯酸酯树脂键合硅胶为填充剂的色谱柱(TSKgel G2500PWxl,7.8 mm×30 cm,7 μm),以磷酸盐缓冲液为流动相对头孢克肟的聚合物进行相关研究[5]。详见表2。
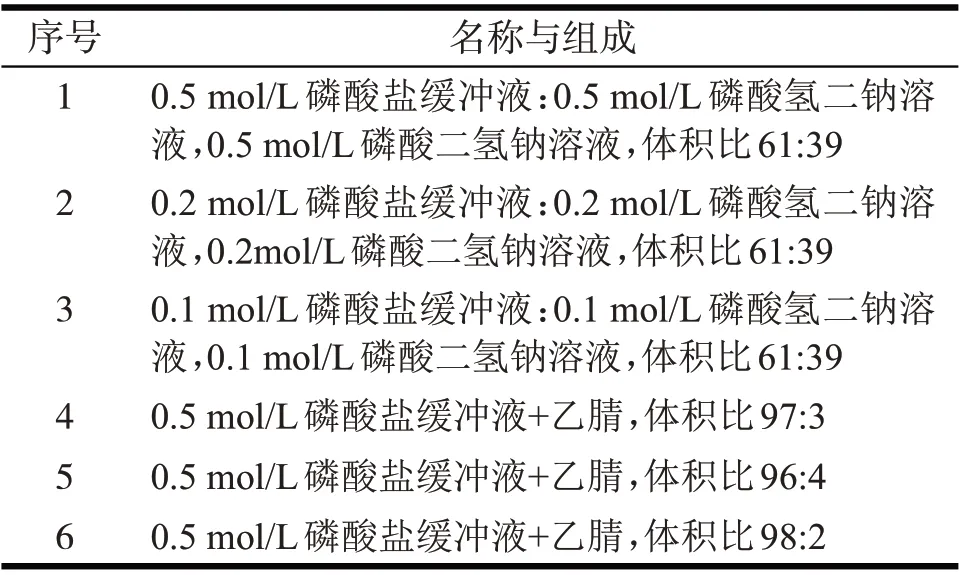
表2 流动相配制Tab 2 Mobile phase preparation
1.5 色谱柱选择
参考文献[7]中头孢克肟高分子杂质的检测条件,采用不同色谱柱、波长对本品(头孢克肟工作对照品)聚合物进行考察。
色谱柱,正相柱,TOSOH TSKgel G2000HHR 30 cm×7.8 mm;色谱柱,反相柱,TSKgel G2500PWxl,7.8 mm×30 cm。流动相DMF+30 mmol/L LiBr+10mmol/L H3PO4(流动相7,DMF为N,N-二甲基甲酰胺),柱温25 ℃,体积流量0.33 mL/min,样品的质量浓度2 g/L,进样量5 μL,检测波长288 nm(根据紫外吸收光谱,288 nm 处为最大吸收拟定)、280 nm(文献条件)、254 nm(同类品种聚合物测定波长)。
1.6 测定方法及限度
精密量取对照品溶液、样品溶液10 μL分别注入液相色谱仪,在系统适用性实验符合上述要求的条件下,记录色谱图。样品溶液的色谱图中如显杂质峰,按外标法以头孢克肟峰计算。除溶剂峰外,所有保留时间小于杂质E峰保留时间的杂质总量不得过1.0%。
2 结果与讨论
2.1 色谱柱选择
按1.4节方法,结果见表3。

表3 色谱柱筛选Tab 3 Column screening
由表2 可知,按文献条件用正相柱在288 nm波长处测定的对照品的聚合物结果,与拟定条件用反相色谱柱在波长288 nm 处测定的头孢克肟工作对照品的聚合物结果基本一致,但杂质个数反相柱更佳。但正相柱条件不适合于考察头孢克肟相关制剂在热水中聚合物的变化情况,且文献条件流动相中DMF的紫外截止波长为268 nm,无法选择254 nm 作为其检测波长,故头孢克肟聚合物的测定未选择文献色谱条件作为检测条件。
2.2 检测波长选择
由样品紫外吸收光谱可知,样品在288 nm 波长处有最大吸收;而同类产品聚合物的检测波长均为254 nm,故比较在2种波长条件下对头孢克肟工作对照品的聚合物进行测定,结果表明,对照品在波长288 nm 处测定的聚合物结果低于在波长254 nm 处测定的聚合物结果,故参考同类品种聚合物测定的波长,选择254 nm 为本品聚合物的检测波长。
2.3 流动相选择
在考察头孢克肟颗粒在热水中聚合物变化情况的实验过程中发现,在热水中降解产生的杂质E峰与主峰以及聚合物峰分离较差,产生的杂质E对聚合物的测定有一定的干扰,故对流动相进行筛选,以排除可能产生的杂质E对本品聚合物测定的干扰。因此,在该实验中对磷酸盐缓冲液及有机相乙腈比例进行考察,结果见表4。
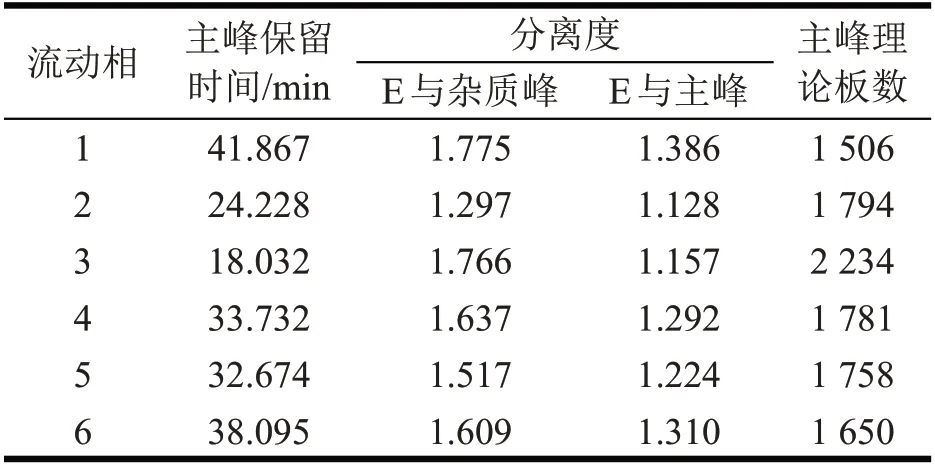
表4 流动相筛选结果Tab 4 Mobile phase screening results
由表4可知,综合考虑主峰保留时间、杂质E与杂质峰分离度、杂质E与主峰分离度、主峰理论板数等相关指标,选择流动相4作为头孢克肟聚合物测定的流动相,其色谱见图1。

图1 流动相4色谱Fig 1 Chromatogram of mobile phase 4
2.4 专属性
2.4.1 空白实验
取空白溶剂(质量分数20%甲醇)过滤,直接进样;取样品空白辅料约5 mg,置25 mL 量瓶中,加5 mL 甲醇振摇溶解5 min,加水稀释至刻度,摇匀,用0.45 μm微孔滤膜滤过,取续滤液10 μL 注入液相色谱仪,记录色谱,结果表明,空白溶剂、空白辅料不干扰本品聚合物测定。
2.4.2 系统适用性
取样品细粉适量(约相当于头孢克肟50 mg),置25 mL量瓶中,加5 mL甲醇振摇溶解5 min,加水稀释至刻度,摇匀,于70 ℃水浴加热30 min,冷却,用0.45 μm微孔滤膜滤过,取续滤液10 μL注入液相色谱仪,记录色谱,结果见表5。

表5 头孢克肟颗粒聚合物系统适用性结果Tab 5 Applicability results of cefixime granules polymer system
由表5可知,杂质E峰(与头孢克肟相对保留时间约为0.89)与头孢克肟峰的分离度应不低于1.0,各成分之间的分离度均符合要求。
2.4.3 杂质定位
头孢克肟特定杂质(杂质A、B、C、D、E、F)在聚合物条件下相对保留时间的确认。
杂质溶液:精密称取头孢克肟杂质A、B、C、D、E、F 约2 mg,分别置25 mL 量瓶中,加5 mL 甲醇溶解,用水稀释至刻度,摇匀,滤过。
蓝色葡聚糖2000 样品溶液:精密称取蓝色葡聚糖2000(SLBW8211)约30 mg,置25 mL 量瓶中,加5 mL 甲醇溶解,用水稀释至刻度,摇匀,滤过,结果见表6。
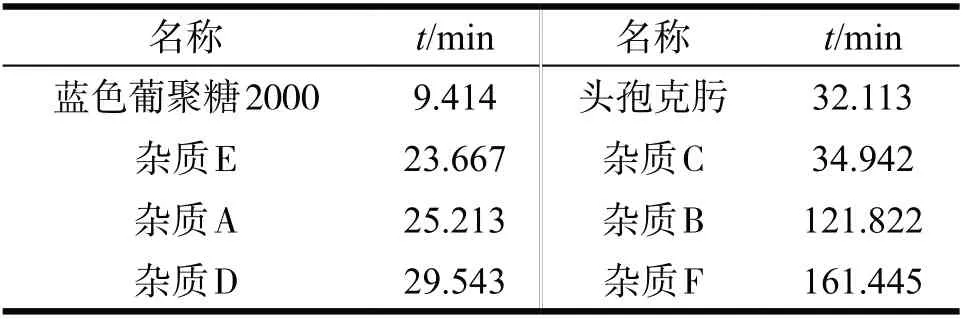
表6 特定杂质的定位研究结果Tab 6 Location study results of specific impurities
由表6可知,在聚合物含量统计中应扣除特定杂质。
2.5 定量限与检测限
聚合物杂质的定量限与检测限以头孢克肟定量限与检测限代替。
按照要求,测定溶液中信噪比(S/N)约为3为检测限,S/N约为10为定量限。
2.5.1 定量限
取头孢克肟工作对照品适量(C16H15N5O7S2的质量浓度4.04 mg/L),精密称定,加甲醇溶解,用水逐步稀释,滤过,进样测定,使头孢克肟峰S/N≈10,作为定量限溶液。进样,计算精密度,结果详见表7(A为峰面积)。
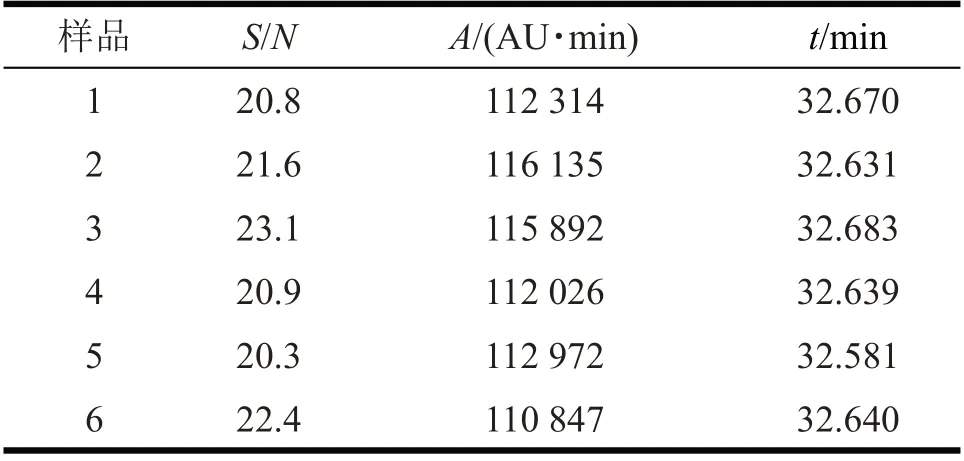
表7 聚合物定量限结果Tab 7 Results of quantitative limits of polymers
由表7 可知,峰面积和保留时间的精密度(RSD)分别为2%和0.11%,符合要求。
2.5.2 检测限
取头孢克肟工作对照品适量(C16H15N5O7S2的质量浓度1.33 mg/L),精密称定,加甲醇溶解,用水逐步稀释,滤过,进样测定,使头孢克肟峰S/N≈3,作为检测限溶液。进样,结果详见表8。
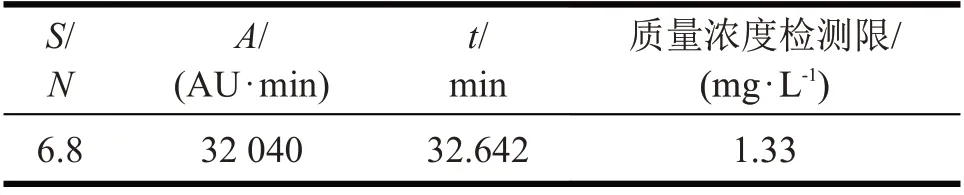
表8 聚合物检测限结果Tab 8 Polymer detection limit results
由表8可知,信噪比大于3,符合要求。
2.6 线性与范围
精密称取头孢克肟对照品114.2 mg,置25 mL量瓶中,加10 mL 甲醇振摇5 min 溶解,加水稀释至刻度,摇匀,作为线性贮备液。取贮备液配制不同含量的线性溶液1~线性溶液6。取线性溶液各10 μL注入液相色谱仪,记录色谱。以浓度和峰面积计算线性方程,结果详见表9。
由表9 可知,拟合线性方程为A/(AU·min)=[3 1017ρ/(mg·L-1)]+353.7,R2=1.000,RSD 为3.4%。头孢克肟(以C16H15N5O7S2计)在质量浓度4.04~2 423 mg/L内与峰面积呈良好线性,符合验证要求,线性回归系数符合≥0.999 的验证要求;y轴(峰面积)截距在测定含量的100%响应量的2.0%以内,符合验证要求。
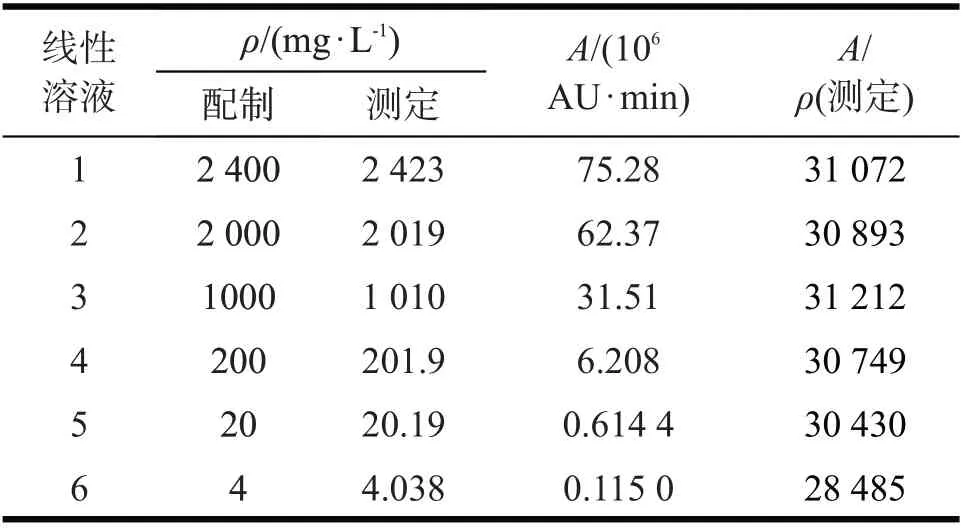
表9 聚合物线性与范围Tab 9 Polymer linearity and range
2.7 精密度
2.7.1 重复性
取头孢克肟对照品约20 mg,精密称定,置50 mL 量瓶,加甲醇5 mL,振摇溶解5 min,用水稀释至刻度,摇匀;再精密量取5 mL,置100 mL量瓶,用水稀释至刻度,摇匀,作为对照品溶液;另精密称取本品适量1.0 g(约相当于头孢克肟50 mg),置25 mL 量瓶中,加甲醇5 mL,振摇溶解5 min,用水稀释至刻度,摇匀,滤过,作为样品溶液,分别配制6份,由1名分析人员进行测定,结果表明,6次测定聚合物的质量分数分别为0.53%、0.53%、0.53%、0.52%、0.53%、0.53%,RSD为0.8%(≤5.0%)重复性良好。
2.7.2 中间精密度
由另一个分析人员使用不同的仪器配制6份相同含量的样品进行测定,结果表明,6次测定聚合物的质量分数分别为0.51%、0.52%、0.51%、0.51%、0.51%、0.51%,RSD为0.8%(≤5.0%),2人所得12 个聚合物结果的RSD 为1.9%(RSD≤5.0%),精密度良好。
2.8 耐用性
2.8.1 溶液稳定性
取头孢克肟聚合物测定样品溶液,室温放置,分别于0、1、2、4、6、8 h 进样,考察聚合物变化情况,结果表明,样品溶液在室温避光条件下放置,聚合物个数一致;聚合物含量逐渐增大,样品溶液应临用新制。
2.8.2 色谱参数变动
取样品溶液,适当的调节柱温、检测波长及流速,考察色谱条件变化后的聚合物结果情况。结果表明:通过考察柱温(±2 ℃)、检测波长(±2 nm)、体积流量(±0.05 mL/min)的改变,样品聚合物个数与聚合物量均无明显差异,本法耐用性良好。
2.9 样品测定结果
3 批 头 孢 克 肟 颗 粒T41181202、T41181203、T41181204,聚合物的质量分数分别为0.094%、0.12%、0.12%。参比制剂(批号DE061)聚合物测定结果为0.086%,均符合限度要求(不超过1.0%)。
3 结 论
通过对不同色谱条件进行筛选,建立TSK 凝胶色谱法测定头孢克肟颗粒聚合物。通过分析方法验证,进行专属性中溶剂空白、辅料空白无干扰,聚合物的质量分数精密度中重复性与中间精密度在0.51%~0.53%,2 组结果RSD 为1.9%<5.0%,在C16H15N5O7S2的质量浓度4.04~2 423 mg/L时线性良好。溶液在常温下放置不稳定,需临用现配,定量限、检测限、耐用性实验结果均符合要求,表明本研究中建立的凝胶色谱法操作简单、专属性强,对头孢克肟颗粒聚合物测定方法提供借鉴。
