氢氧根交换膜燃料电池阳极氢氧化反应中Ru基电催化剂的进展*
2021-02-15孟凡超王海斌孟祥智胡冠群李春雷丛媛媛赵秋萍
孟凡超,王海斌,孟祥智,胡冠群,李春雷,丛媛媛,赵秋萍
(兰州理工大学 石油化工学院,甘肃 兰州 730050)
在过去的几年里,人们对于能源的需求不断增加,导致了化石燃料的消耗不断增加,环境污染和气候变化问题也随之而来。这些都在迫使人们去探索更加清洁环保的可再生能源。在各种新能源中,由于氢能源有着零污染和能量密度高的特点,使得氢能源备受关注并被认为是最有前途的能源之一。燃料电池可以将化学能直接转化为电能,且该过程不受卡诺循环的影响,具有清洁和能量转换效率高等优势[1]。随着质子交换膜燃料电池(PEMFCs)技术飞速进步,氢燃料电池汽车一进入市场就大受好评[2]。但是目前的PEMFCs严重依赖于Pt基电催化剂,这限制了其商业化发展。氢氧根交换膜燃料电池(HEMFCs),又称碱性聚合物电解质燃料电池[3](APEFCs),其由PEMFCs衍生而来,使用主链上接有阳离子用来传递OH-的聚合物电解质膜替代用来传递H+的全氟磺酸膜,将酸性环境转变为碱性环境。就目前而言,HEMFCs也面临着和PEMFCs同样的困境,那就是依赖Pt而成本高昂。Pt及其合金仍然是HEMFCs氧还原反应(ORR)和氢氧化反应(HOR)的最佳电催化剂[4]。对于HEMFCs阴极ORR,目前已研发出一些性能优异的非贵金属电催化剂,如Fe-N-C、Co-N-C等可以替代Pt基ORR电催化剂,大大降低燃料电池阴极侧的成本[5]。为了使HEMFCs性能可以与PEMFCs相媲美,需要在阳极侧喷涂较高载量的Pt。这是因为当电解质由酸性变为碱性时,Pt的碱性HOR活性降低了近100倍,需提高Pt的载量以加速反应速率[6-7]。阳极侧成本的提高削弱了阴极低成本的优势。因此,开发高效、稳定、低成本的低Pt或无Pt碱性HOR电催化剂对于HEMFCs的发展至关重要[8-9]。
尽管在过去的几年里陆续报道了非贵金属的碱性HOR电催化剂,但是其催化活性以及稳定性依然逊色于贵金属电催化剂[10]。近年来,研究学者们发现Ru具有丰富的氧化还原电化学性质,且价格仅为Pt的6%~36%,存在着明显的成本优势,是替代Pt的最佳候选者。纯金属Ru,作为碱性HOR电催化剂,因其本身活性不高且在高的阳极电势下容易发生氧化[11]。因此围绕引入其他组分,调变Ru的电子分布,提高Ru基电催化剂的活性,开展了相关工作。
性能优异的Ru基电催化剂的制备依赖于碱性HOR机理的认识。作者阐述了在碱性介质中HOR的电催化机理,说明了影响HOR活性的因素,在此基础上总结了几十年来阳极Ru基电催化剂的技术进展并对未来的研究方向提出了展望。
1 碱性介质中HOR的作用机理
在碱性介质中,HOR反应过程可以分为以下3个步骤。

(1)
(2)
(3)
式中,Had表示吸附态的氢,*表示电催化剂的活性位点[12]。
反应速率决速步骤(RDS)是一系列化学反应当中速率最慢的步骤,改善RDS的速率有利于提高整个反应过程的速率[12]。Shinagawa等[13]根据RDS的不同将碱性HOR反应历程分为Tafel(RDS)-Volmer、Tafel-Volmer(RDS)、Heyrovsky(RDS)-Volmer和Heyrovsky-Volmer(RDS)4类。
对于碱性HOR性能评价而言,存在着2个活性描述符,一个是氢键能(HBE),另一个是亲氧性(OHBE)。研究学者们对于2个活性描述符在反应中仅氢键能起决定作用还是共同作用还没有达成统一(见图1)[14]。
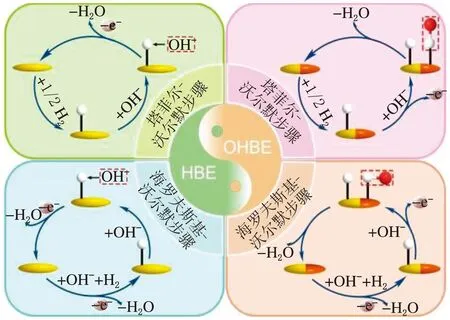
图1 碱性HOR中HBE和OHBE的理论示意图
一种观点认为,反应的活性只与氢键能有关。通过Sabatier[15-17]规则可知,电催化剂对H的吸附不能太强也不能太弱,当吸附过强时中间物质的脱附就会变得困难,然而当吸附太弱时,H的覆盖度就会不足。Sheng等[18]赞同HBE理论,认为该理论与在酸性介质中相同,Had是HOR过程中唯一的中间吸附物质,如果能够达到最佳的HBE就可以保持Had的吸附和脱附平衡。因此提出可以通过改变条件达到最优的HBE从而实现催化活性的最优化。Yan等[19]发现,Pt的HBE随着电解质pH值的增加而增加,在碱性条件下,Pt的HBE偏离理想状况,使得HOR活性降低,故其认为HBE是HOR的唯一活性描述符。Ohyama[20]课题组探究了Ru3Ir2/C和RuIr/C在碱性溶液中的HOR活性,研究结果表明在碱性介质中Ru3Ir2/C的活性要优越于RuIr/C,其主要原因是因为Ru3Ir2/C的合金化使得其HBE相较于RuIr/C降低了。
另一种观点是HBE和亲氧性共同影响碱性HOR活性。Markovic[21]课题组在研究中发现,用Ni(OH)2修饰Pt单晶,其在碱性中的活性会提升大约8倍,将这种活性的提升归功于亲氧组分的引入有利于捕捉含氧物种。Li等[22]在溶液中加入RuCl3时,Pt/C的活性会大大提高,认为通过电位扫描,电催化剂表面引入了亲氧性强的组分,提高了电催化剂的碱性HOR活性。此外,Strmcnik等[23]也支持随着亲氧位点的增加,电催化剂的碱性HOR活性也会随之增加。Duan[24]课题组的研究发现MoNi4相较于纯Ni电催化剂,其活性显著增加,这是因为Ni和Mo的合金化降低了HBE,与此同时引入的Mo增强了亲氧性,大大促进了Volmer步骤,提高了碱性HOR活性。Alesker等[25]将Pd和Ni的混合纳米颗粒(NPs)用作HEMFC的碱性HOR电催化剂,其峰值功率密度(PPD)为0.40 W/cm2是纯Pd(0.18 W/cm2)电催化剂的2.2倍。提出在不考虑合金化和电子效应的影响下,NPs中Ni的亲氧性是提升HOR活性的关键所在。Liu等[26]通过原位X射线吸收谱(XAS)观察到了在HOR的电位区间内存在OHad,并进一步提出了OHad通过双功能机理促进了Volmer步骤进而促进了碱性HOR反应。Ishikawa[27]课题组合成了Ru-Ir/C电催化剂,通过测量其Tafel斜率发现Volmer步骤是HOR的RDS,同时Tafel步骤也在影响着整个反应,因此提出了碱性HOR反应在Ru-Ir电催化剂表面上通过双功能机理而得以增强。当然也有学者对这个说法提出了异议,称并非所有的条件下都适用于此机理,比如Ramaswsamy等[28]就提出了在Pt与Nb、Ni、Cu等过渡金属形成的合金表面,在高pH值条件下不是形成Hupd而是一层氧化物,此时才涉及到HOR双功能机理,即Pt-Had与过渡金属氧化物上形成的OHad直接发生反应。Liu等[29]同样发现只有约0.9 Vvs相对于标准氢电极(vs. RHE)时,OHad具有更好的稳定性,更可能参与到反应中,这时便遵循双功能机理。
2 Ru基电催化剂
Ru金属价格低廉,是目前有潜力替代Pt的阳极电催化剂,吸引了越来越多的学者。然而,Ru的碱性HOR活性一直低于Pt,这是由于Ru具有较强的氢键能以及亲氧性[11],因此需要引入其他组分,优化电催化剂的氢键能或亲氧性,以提高电催化剂的活性。
2.1 Ru-M合金电催化剂
通过Ru金属与其他金属形成合金作为燃料电池的电催化剂,其碱性HOR电催化剂的活性以及稳定性可以得到显著提高,因为合金组成以及晶体结构的调变会改变电催化剂中Ru的电子结构,从而优化电催化剂的氢键能或亲氧性,提高碱性HOR活性。
Papandrew[30]等人基于化学气相沉积工艺合成了分散在碳载体上的Ru基RuxMy(M=Pt或Pd)合金电催化剂,表征结果显示决速步骤取决于合金材料的选择,在合成的一系列合金化合物中Ru0.20Pt0.80和Ru0.20Pd0.80表现出最佳活性,碳担载的Ru0.20Pt0.80和Ru0.20Pd0.80电催化剂的HOR面积比交换电流密度(j0,s)分别是1.42和0.148 mA/cm2,是Pt/C(0.49 mA/cm2)和Pd/C(0.05 mA/cm2)的3倍。
Ishikawa[27]等人通过多元醇法合成了碳担载的不同金属比例的Ru-Ir合金纳米颗粒电催化剂(Ru-Ir/C),与Ru/C和Ir/C相比,所有Ru-Ir/C合金电催化剂均表现出更高的活性,这表明合金化可以有效提高碱性HOR活性。当Ru/Ir的原子比为2∶3时,其j0,s大约为0.53 mA/cm2,分别是纯Ru和纯Ir电催化剂的19.5倍和3.5倍。
Scofield[31]团队合成了Pt7M3合金纳米线(NWs,M=Ru、Fe、Co、Cu、Au),在测试中发现Pt7Ru3NWs表现出最高的HOR活性,在0.05 V vs.RHE条件下表现出了2.2 mA/cm2的活性以及0.493 mA/cm2的面积比活性,优异于纯Pt NWs(1.38 mA/cm2和0.229 mA/cm2)。
Cong[32]等人通过湿法浸渍,高温还原和高温NH3蚀刻合成了负载在氮掺杂碳(N-C)上的一系列Pt1-xRux颗粒(见图2)[31],在一系列的合成物中Pt0.25Ru0.75/N-C表现出了最高的碱性氢氧化活性,其质量比交换电流密度(j0,m)为1 654 A/g PtRu,分别是商业Pt/C(352 A/gPt)和PtRu/C(1 213 A/g PtRu)的4.7倍和1.4倍。
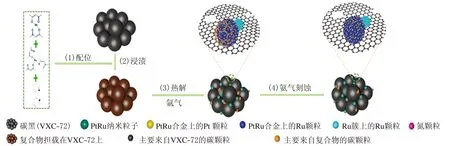
图2 Pt0.25Ru0.75/N-C电催化剂的合成示意图
Wang[33]的课题组为了研究合金化对于催化活性的影响,合成了Ru-M/C(M=Co、Ni、Fe)电催化剂,在实验过程中发现随着Co、Ni或Fe含量的增加,电催化剂H的吸附/解离过程的可逆性增强使得HOR活性也随之增强。其所有的Ru0.95M0.05/C(M=Co、Ni、Fe)电催化剂的活性均高于Ru/C,有的甚至高于Pt/C。Ru0.95Co0.05/C的活性要略高于Ru0.95Ni0.05/C和Ru0.95Fe0.05/C,其j0,m是纯Ru的5倍。
Xue[34]等人报道了一种无铂电催化剂Ru7Ni3/C,与先前报道的Ru基电催化剂相比,其具有优异的碱性HOR活性,在0.05 V vs.RHE处的质量比活性为9.4 A/mgRu,分别为Ru/C(0.28 A/mgRu)、Pt/C(0.45 A/mgPt)和PtRu/C(3.5 A/mgPt+Ru)的33、21和3倍。在0.05 V vs.RHE处的面积比活性为23.4 mA/cm2,分别是Ru/C(0.67 mA/cm2)、Pt/C(0.93 mA/cm2)和PtRu/C(4.8 mA/cm2)的35、25和5倍(见图3)[34]。如此优异的性能要归功于Ni与Ru的合金化作用以及表面氧化的Ni物种共同削弱了Ru-H键强。
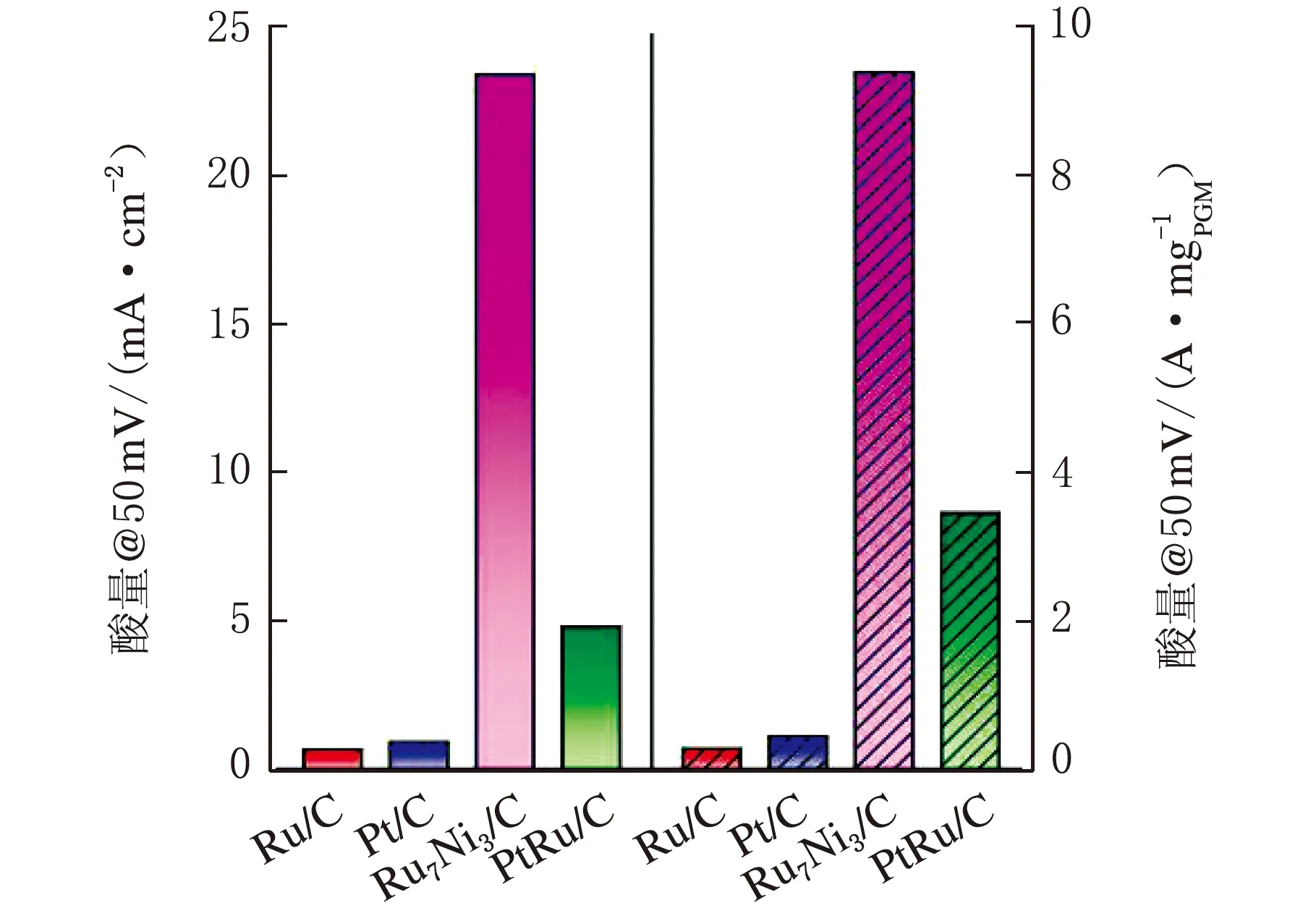
图3 Ru/C、Pt/C、Ru7Ni3/C和PtRu/C电催化剂在50 mV vs.RHE处的质量比活性和面积比活性对比图
基于上述的研究表明,Ru与其他金属的合金化可以有效地调变Ru的电子结构,从而有效提高了Ru基电催化剂的碱性HOR活性。
2.2 核壳结构Ru基电催化剂
为了能够实现减少电催化剂中贵金属的负载量,以降低成本的同时还能保持高的电催化活性和稳定性,研究者们提出了核壳结构电催化剂。核壳结构纳米颗粒是由至少2种相同或不同的物质组成的复合颗粒,其中一种物质成为核,另外一种物质形成包裹在外面的壳[35]。特殊的颗粒结构能够很好地保留核心金属原有的性能,同时还可以具有外层金属的特性。这种复合材料能够很好地克服单一金属材料固有的缺陷,使得电催化性能和稳定性得到很大地提升,同时也能有效地解决贵金属负载量高导致的成本问题。
Xing[36]课题组提出使用欠电位沉积(UPD)方法在Ru纳米颗粒上逐层生长Pt层来调节表面性能,经过重复的UPD工艺获得Ru NPs上的Pt多层结构,以此形成以Ru为核、Pt为壳层的核壳结构电催化剂。从Butler-Volmer图上可以获得Ru-rGO、Ru-MC(Pt原子单层沉积在Ru NPs) Pt-rGO、Ru-DC(双层) Pt-rGO、Ru-TC(三层) Pt-rGO和Ru-QC(四层) Pt-rGO电催化剂的j0,s值分别为0.27、1.49、2.41、1.76和1.67 mA/cm2。显然,所有合成的Ru@Pt核壳电催化剂都有着比Ru-rGO更好的电化学活性。运用循环伏安法(CV)研究M-H键能,结果表明所制备的一系列核壳结构电催化剂的UPD-Hdes的峰位置较Pt/C发生了负移,表明Ru能够降低Pt-H的键能,也在某种程度上解释了Ru@Pt核壳NPs优异的电催化性能。同时选用了CO溶出伏安法来探究亲氧性,结果发现Ru-TC Pt-rGO和Ru-QC Pt-rGO上的CO溶出峰随着Ru核上Pt厚度的增加而正向移动,交换电流密度几乎随表面亲氧性的降低而线性降低。基于这2个结果提出HOR活性是通过双功能机理实现的。其中Ru提供了吸附OH的活性位点,而附近的Pt原子为Had提供了活性位点而且Pt壳原子与Ru核原子之间的电子相互作用也可能增强了HOR活性。
2.3 Ru-金属氧化物电催化剂
近几年研究者们把目光逐渐转移到了金属-金属氧化物电催化剂上,因为部分金属氧化物在高电位、碱性条件下具有良好的稳定性。此外,金属氧化物与金属电催化剂之间强的相互作用可以在一定程度上改变金属表面的电子结构从而改变金属电催化剂的电催化活性,提高金属电催化剂的稳定性。
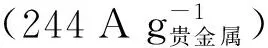
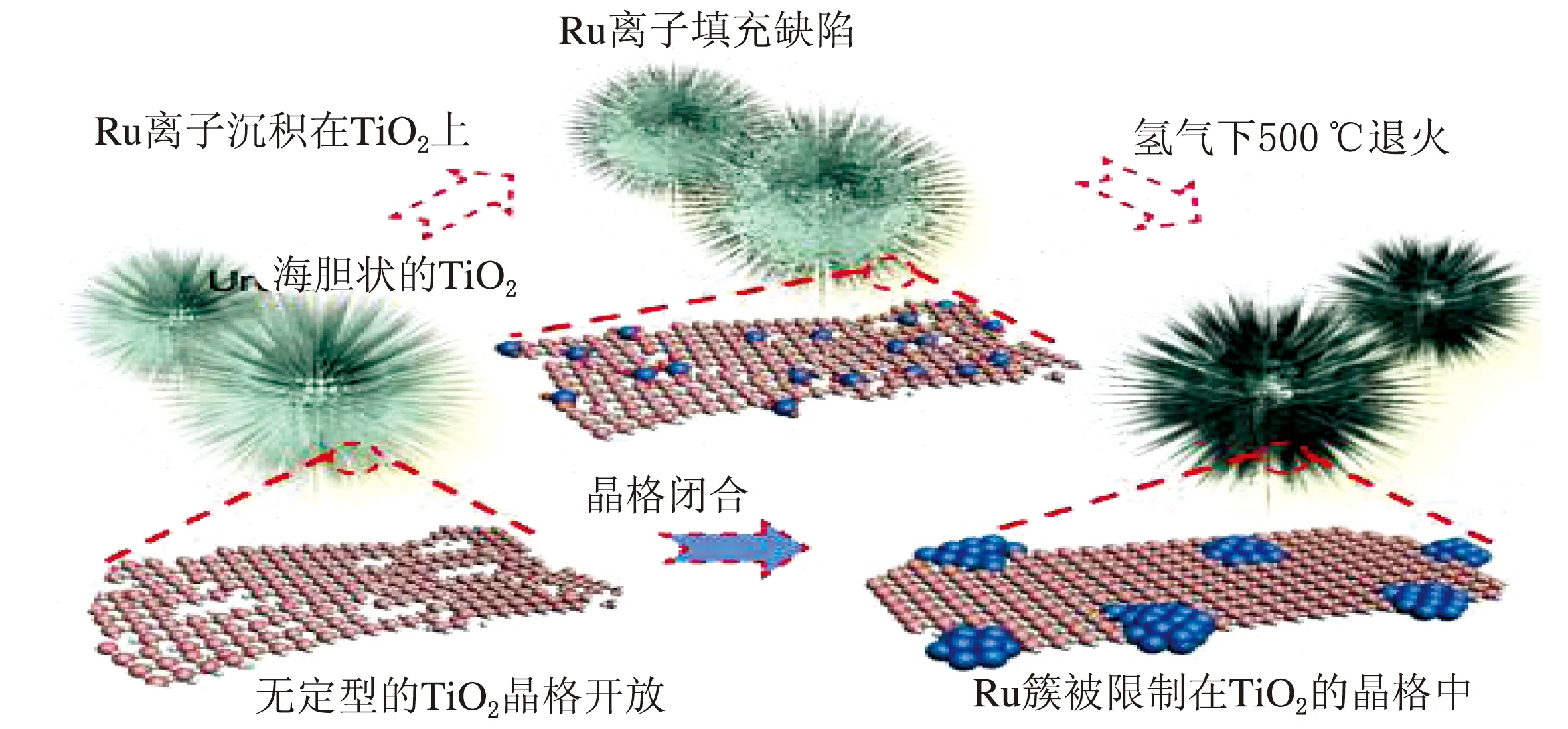
图4 Ru@TiO2电催化剂的合成示意图[37]
Jiang[38]等提出了相间氧化的概念(见图5),Ru被TiO2相间氧化,2 nm的Ru簇可以在高达0.9 V(vs.RHE)的电位下有效地催化HOR,而自身不会被过度氧化。所制备的相间氧化电催化剂IO-Ru-TiO2/C的质量比活性为907 A/gRu@0.05 V vs.RHE,分别是Ru/C和PtRu/C电催化剂的17.5和1.5倍,其面积比活性为1.13 mA/cm2,分别是Ru/C(0.12 mA/cm2)和Ru-TiO2/C(0.15 mA/cm2)近9.4倍和7.5倍。
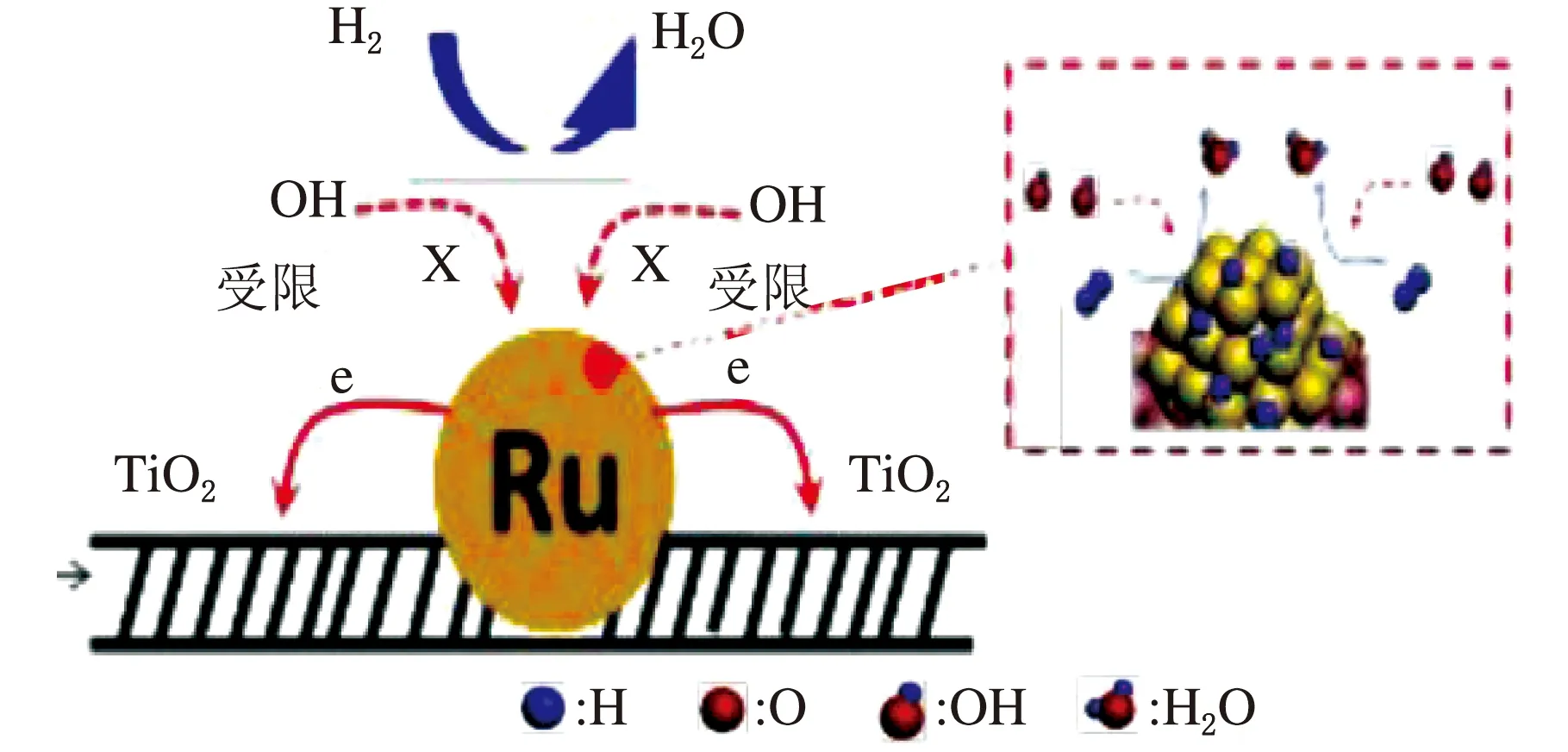
图5 IO-Ru-TiO2/C相间氧化电催化剂的合成示意图
活性的提高要归因于形成了丰富的Ru-TiO2界面,界面上产生的电子转移将Ru簇氧化为近似Ru4+,从而形成了相对稳定的半填充的4d轨道。这就使得Ru的进一步氧化变得困难从而保留了金属表面。
2.4 载体优化的Ru基电催化剂
一般来说,载体本身是不具有电催化性能的,载体主要用于支持活性组分,使电催化剂具有特定的物理性质。合适的载体能够使制得的电催化剂具有合适的形貌、尺寸等,也可以使电催化剂活性组分均匀分散在载体表面,以此获得更高的电化学活性比表面积,提高单位质量电催化剂的催化效率[39]。载体的不同可以使电催化剂的性能有很大的差异,因此对于目前来说优化电催化剂的载体也是解决目前电催化领域问题的方法之一。
Zeng等[40]设计了一种具有中孔沟状通道的介孔碳载体(Meso C)负载金属Ru电催化剂。这种优化了载体结构的Ru/Meso C电催化剂有着十分优异的碱性HOR活性,其质量比活性为0.54 mA/μgRu是Ru/C(0.19 mA/μgRu)的2.8倍,其峰值功率密度为1.02 W/cm2,是Ru/C(0.76 W/cm2)的大约1.3倍,甚至可以在相同金属载量下与商用Pt/C(20wt%)的单池性能相媲美。其课题组是为数不多的通过设计亲水和疏水界面优化碱性HOR电催化剂的性能,所涉及的介孔碳载体有着中孔微观结构,一方面能够形成外部亲水且内部疏水表面,从而确保了H2吸附和解离的理想界面环境,促进了氢的氧化。另一方面,多孔通道可以充当密闭空间,以防止内部预先锚定的Ru NPs过度暴露于空气,从而保留比Ru/C更高比例的Ru金属态。这种有效提高HOR活性降低贵金属负载量的方式也为日后电催化剂设计提供了新思路。
Brkovic等[41]结合氧化钨和碳化钨的优点设计出了一种新型的电催化剂载体——非化学计量的碳化钨氧化物(WxCyOz),主要是由非化学计量的氧化钨(WOx)和碳化钨(WC)组合而成,将其用作Pt-Ru纳米颗粒的载体。在研究中发现质量分数为30%金属载量的Pt-Ru/WxCyOz有着比商业Pt/C更好的电催化活性,在30 mV的HOR过电位下,质量分数为30%的Pt-Ru/WxCyOz表现出8.33 mA/cm2的动力学电流密度(jk)和0.028 A/mgPt的质量比活性,这些性能均优异于商业Pt/C(7.86 mA/cm2和0.197 A/mgPt)。
3 结束语
首先阐述了碱性HOR的作用机理,随后着重总结了如今运用于碱性HOR的Ru基电催化剂,主要包括了Ru-M合金电催化剂、核壳结构Ru基电催化剂、Ru-金属氧化物电催化剂以及载体优化的Ru基电催化剂。尽管近几年来碱性HOR电催化剂的研究取得了很大的进展,但是就目前来说依然面临着诸多挑战。
HBE作为碱性HOR的重要指标之一,其很难在电化学条件下直接获得,因为不是所有的金属都具有不受干扰的氢吸附/脱附区,部分金属的氢区涉及OH-的吸附。此外,OH-的作用也有待商榷,因此需要其他更实际有效的光谱方法在电催化条件下分析电催化剂上的HBE以及亲氧性与活性之间的关系。
尽管使用Ru基电催化剂能够降低阳极电催化剂的成本,但是Ru容易氧化,在电位较高的情况下,吸附的含氧物种会抑制氢的吸附,降低碱性HOR活性。Xue[42]等人曾在其文章中指出在实际应用时,HEMFCs可能会出现偏离正常阳极工作条件的情况,比如电池启动和关闭时,阳极会暴露在较高的电位下同时可能会出现反向电流,这就对阳极电催化剂在高电位条件下仍需具有稳定高效催化HOR有了更高的要求。
为了更好地研究和设计碱性HOR电催化剂,可以使用原位技术实时观测碱性HOR的催化过程,探索碱性HOR反应中间体,这有助于明确电催化剂的真实催化活性位点,为开发高性能的电催化剂提供理论指导[14]。此外,可以借助密度泛函理论认识电催化剂活性位点的最优结构,进一步优化电催化剂的制备参数,解决高电位条件下Ru易氧化问题,构筑具有高交叉频率、高稳定性活性位点的Ru基碱性HOR电催化剂。
