G蛋白偶联受体的共同激活机制
2021-02-07周庆同戴之卓赵素文
周庆同,戴之卓,赵素文†
①复旦大学 基础医学院,上海 200032;②上海科技大学 iHuman研究所,上海 201210;③上海科技大学 生命科学与技术学院,上海 201210
G蛋白偶联受体(G protein-coupled receptor,GPCR)是人体内最大的一类细胞信号转导受体家族,几乎参与所有生命活动,负责调控细胞对光线、气味、激素、神经递质、趋化因子等的应答,对维持生命健康具有非常重要的意义[1-2]。GPCR的重要性使其成为最重要的药物靶标家族之一,在心血管疾病、代谢性疾病、神经相关性疾病、免疫性疾病和癌症等重大疾病的药物研发中扮演着重要的角色。目前靶向GPCR的药物约500个,占FDA已批准药物的34%[3-4]。基于序列相似性,人类的800多个GPCR可被分为4个亚家族,分别是视紫红质受体(A家族)、分泌素受体和黏附受体(B家族)、代谢型谷氨酸受体(C家族)、卷曲受体(F家族)。其中,A家族的受体个数最多(超过700个),生理功能也最为多样。目前GPCR研究已获得10次诺贝尔奖,这也从侧面说明它的重要性[5]。
GPCR具有保守的结构特征,即7个跨膜螺旋(seven transmembrane helices, 7TMs),这7个螺旋组成的跨膜结构域将受体分割为胞外N端(N-terminus)、胞内C端(C-terminus)、3个胞外环(extracellular loops)和3个胞内环(intracellular loops)。GPCR的胞外区域在结合激动性信号分子(如气味、激素、神经递质、趋化因子)后发生构象变化,进而引发跨膜螺旋的运动,特别是第6跨膜螺旋靠近胞内的那部分(intracellular half of TM6)往外翘起。此时激活受体的胞内区域可招募并结合下游效应蛋白(如G蛋白、β-arrestin等),这些效应蛋白通过环腺苷酸(cAMP)信号通路、磷脂酰肌醇信号通路和钙离子信号通路等调节体内生理活动。
GPCR的激活机制是理解其生理功能及相关疾病成因的必由之路,也是药物精准研发的基础,并一直是该领域研究的核心内容和前沿热点[6-7]。对GPCR的激活机制研究多关注于单个受体或个别位点,所揭示的激活机制缺乏家族层面的系统总结。目前,伴随着新的激活现象和特征的发现,人们对受体激活机制的传统认识正不断被突破。
1 GPCR激活机制研究中的方法与创新
GPCR的激活是指激动剂结合引发的下游效应蛋白的招募。GPCR激活的本质是胞外侧激动剂结合和胞内侧下游效应蛋白招募的互相耦合、别构通信的过程,即受体从非激活态到激活态的构象转变(图1)。因此,研究GPCR激活机制不仅需要高精度的GPCR三维结构,而且需要精确描述GPCR构象变化的分析工具。科学家经过坚持不懈地努力,对GPCR特别是代表性受体(如β2肾上腺素受体、视紫红质受体、A2A腺苷受体)的激活过程或方式的理解日益深入,但对家族层面的共同激活机制却知之甚少。究其原因,一方面是结构被解析的受体数量仍然有限,另一方面是缺乏可精确描述构象变化的分析工具,显然仅依靠肉眼观察或位移测量等传统方法无法做到准确和系统地研究。以下重点阐述GPCR激活机制研究中的主要方法(表1)与创新点。

图1 大麻素受体CB1的构象变化(左侧为结合拮抗剂时处于非激活时的CB1结构[8];右侧为结合激动剂和G蛋白时处于完全激活态的CB1结构[9])

表1 GPCR激活机制的研究方法
1.1 三维结构测定
由于GPCR的表达、纯化和结晶都存在极大的困难,GPCR的三维结构一直很难测定。伴随着GPCR昆虫表达系统的建立、融合蛋白和热稳定性突变等的引入、高亲和力配体的筛选使用、脂立方相(lipidic cubic phase, LCP)膜蛋白结晶技术的发展[10],GPCR的X-射线晶体学研究已取得巨大成功。其中代表性的成就有2007年Brian Kobilka和Raymond Stevens等[11]合作解析的第一个人源GPCR-β2肾上腺素受体(β2adrenergic receptor, β2AR)晶体结构,2011年Brian Kobilka等[12]解析的第一个GPCR-G蛋白异源三聚体晶体结构和2015年徐华强等[13]解析的第一个GPCR-βarrestin复合物晶体结构。2017年冷冻电镜技术被应用到GPCR的结构解析中[14-15],使GPCR的三维结构测定变得容易。值得欣喜的是,近年来多个GPCR的结构首先被我国科学家解析[8,13,16-26]。
截至2020年9月,GPCR已有超过80个受体的400余个结构被解析,其中A家族有近60个受体的300多个结构被解析。这些结构揭示出各受体在配体结合模式、耦合的G蛋白亚型、激活方式等方面具有明显差异。依据配体分子的药理学特性和受体的整体状态,这些结构可分为3类:①拮抗剂或反向激动剂结合的非激活态(inactive state);②同时结合激动剂和下游效应蛋白(如G蛋白、β-arrestin等)的激活态(active state);③只结合激动剂的中间态(intermediate state)。A家族有多个受体处于非激活态和激活态时的结构都已被解析,如牛视紫红质受体(bRho)、β2肾上腺素受体(β2AR)、 M2毒蕈碱型乙酰胆碱受体(M2R)、μ阿片受体(μOR)、 A2A腺苷受体(adenosine A2Areceptor, A2AR)和κ阿片受体(κ-OR),为GPCR家族层面的共同激活机制研究提供了良好起点。
1.2 核磁共振和分子动力学模拟
与三维结构测定的稳定的静态构象不同,核磁共振(nuclear magnetic resonance, NMR)能够在原子分辨率水平研究蛋白质的构象变化,填补了晶体或电镜结构缺失的信息,为理解GPCR动态构象变化和激活机制提供全新的视角。NMR在GPCR应用上的主要挑战是信号弱且杂,设计新颖有效的标记位点颇为耗时耗力。由于GPCR分子较大,NMR只能使用15N选择性标记一两种出现频率不高且结构分布较广的残基(如Gly和Trp)[27],或者使用含有19F的分子来反应性地标记事先选定的与激活关系紧密的一两个Cys位点[28-30]。随着高场NMR、改进的低温探针、稳定同位素标记及横向弛豫优化谱(transverse relaxation-optimized spectroscopy, TROSY)等技术的应用,GPCR的NMR研究取得突破性发展,如鉴定出GPCR存在着多种与功能密切相关的构象状态,且受激动剂、拮抗剂、别构调节剂和下游效应蛋白等调控[27-28,31]。
借助于不断优化的分子力场和计算机软硬件,分子动力学模拟已被广泛应用于生物大分子的结构和动态特性研究,在GPCR研究中正发挥着日益重要的作用[32-33]。分子动力学模拟使人们得以了解药物分子如何进入结合口袋并激活受体,跨膜信号如何转导,药物的偏向性信号通路如何实现,胆固醇等脂质分子如何影响受体激活等重要问题,也为GPCR的药物设计和激活机制研究提供重要的研究工具。GPCR嵌膜模拟体系通常含原子数较多,模拟时间一般在微秒水平,无法覆盖GPCR的整个激活过程,且不同受体的激活过程也会略有差异,因此通过分子动力学模拟研究家族层面的GPCR激活机制仍有较大困难。
1.3 残基接触
GPCR激活的本质是局部残基间相互作用的改变诱发螺旋位置的变化,因此有必要开发计算方法来描述残基间相互作用从非激活态到激活态的转变,进而比较各受体在激活时的残基作用改变的异同,最终建立家族层面的共同激活机制。2016年英国剑桥MRC-MLB的Madan Babu课题组,使用残基接触(residue contact, RC)来研究GPCR的激活机制。残基接触的定义是:若两残基有原子对间距离减去其范德华半径之和小于0.5 Å,即认为两残基间存在接触,否则两残基间没有接触[34]。对A家族GPCR中同时有激活和非激活态结构的5个受体10个结构进行残基接触分析,研究人员发现有6组残基对在这5个受体激活过程中具有一致的残基接触变化,即同时消失或形成。其中,2组残基对(3×46—7×53和5×55—6×41)在激活后新形成接触,余下的4组残基对(1×53—7×53,3×46—6×37,7×53—8×50和7×54—8×51)则在激活后接触消失。这里残基编号采用GPCRdb命名法[35]:将每个α螺旋上序列最保守的残基定为×50,其他残基依据与×50在序列上的相对位置依次命名,如3×49即为第3个α螺旋上、在序列上位于最保守位点3×50的前一位,而3×51则为第3个α螺旋上、在序列上位于最保守位点3×50的后一位。结合A家族其他结构的验证,他们发现靠近G蛋白结合区域的两组残基对(3×46—6×37和3×46—7×53)具有家族普遍性,即A家族GPCR激活时都有这样的构象接触变化,因此A家族GPCR被认为具有多样化的激活通路,且这些激活通路仅收敛于G蛋白结合区域。这一工作为GPCR激活机制研究提供了全新的计算思路,但其揭示的共同激活机制却遗漏了一系列保守的特征基序(motif,序列中局部的保守区域),如CWxP[27,36]、PIF[37-38]、DRY[39-40]、钠离子结合口袋等[27,41],也无法解释TM6末端因何翘起,因此GPCR的共同激活机制仍有待于完善。
受到配体-残基相互作用定量描述的启发[42],我们提出残基接触打分(residue-residue contact score, RRCS)用以精确描述残基对间的接触强度[43],将三维的、复杂的结构信息转化为二维的、量化的残基接触打分,同时残基接触分差(ΔRRCS = RRCSactive-RRCSinactive)可精确描述受体激活过程中的残基构象变化。残基接触打分完全从蛋白质结构本身出发,不依赖于蛋白质结构比对(alignment),且可将整体和局部、显著和微小的构象变化系统地划分为开关(switching)和重排(repacking)、螺旋间(inter-helical) 和螺旋内(intra-helical)等两个层面的4种类型。为遴选出A家族GPCR激活时都具有的保守的构象变化,我们首先分析6个受体(bRho、β2AR、M2R、μOR、A2AR和κ-OR)在激活过程中的残基接触分差。一方面,检查这些残基对在6个受体中是否有统一的构象变化,即残基接触分差皆为正或负;另一方面,研究这些残基对的接触打分,在家族层面的非激活与激活态间是否存在显著性差异(142个非激活态结构和27个激活态结构)。我们最终发现在A家族GPCR激活时,有34组残基对具有保守的构象变化。
2 GPCR的共同激活机制
2.1 A家族GPCR的共同激活机制
基于家族保守的、激活时具有统一构象变化的34组残基对,我们构建起A家族GPCR的共同激活通路(图2)。这34组残基对由35个残基构成,有机连接和整合了人们熟知的、序列保守但空间疏离的特征基序,如CWxP[27,36]、PIF[37-38]、DRY[39-40]、钠离子结合口袋等[27,41]和NPxxY[12,34]等,在残基水平上回答GPCR如何被激活,TM6为什么会翘起,各基序如何协同作用实现GPCR的跨膜信号转导。需要指出的是,这一通路是A家族不同受体在不同激动剂或不同下游效应蛋白时的共同部分,每个受体仍然具有独特的、受体/配体/效应蛋白特殊的激活通路。
依据这35个残基的空间位置和功能,我们将GPCR的共同激活通路由胞外到胞内共划分为4层(图2(a)):①信号启动层;②疏水锁层;③微型开关传递层;④胞内效应蛋白结合层。激动剂的结合诱发配体结合口袋发生构象变化,最终引起位于胞内口袋底部的特征基序CWxP、PIF发生结构重排,使得钠离子结合口袋坍塌,最终实现信号启动。第二层疏水锁层残基在接收到第一层的启动信号后,几个疏水残基构成的疏水锁(6×41—3×43 和 6×40—3×43)被打开,分别通过TM6和TM7传递激活信号。第三层由2个微型开关残基(6×37和7×53)控制,在接受到疏水锁层的信号后发生剧烈的构象变化,形成或破坏了一系列的残基接触使得信号得以放大。第四层则将上述构象变化通过结合胞内效应蛋白传递到胞内,最终通过第二信使cAMP、IP3和Ca2+等实现信号转导。综上,4层残基互相配合,通过螺旋间/螺旋内的残基开关或重排实现GPCR的跨膜信号转导。
为表征跨膜螺旋的整体构象变化,我们选择TM3和TM6间的所有4组残基对来计算2个螺旋间的接触分,即RRCSTM3-TM6,类似地还有RRCSTM3-TM7和RRCSTM5-TM6。在将142个非激活态结构和27个激活态结构投影到由RRCSTM3-TM6和RRCSTM3-TM7组成的二维空间时,我们发现GPCR的激活态和非激活态具有迥然不同的结构特征(图2(b))。激活态的RRCSTM3-TM6为零,而RRCSTM3-TM7通常大于5;非激活态的RRCSTM3-TM7通常接近或等于零,而RRCSTM3-TM6分布广泛但通常大于零。这充分说明GPCR激活时具有共同的整体构象变化,即激动剂的结合触发TM6胞内段往外运动从而远离TM3,这使得TM7可以往内运动从而靠近TM3,最终在受体的胞内侧形成空隙以结合G蛋白等下游效应蛋白。值得注意的是,此前研究者在使用肉眼观察受体结构来判断激活状态时,常过分关注TM6翘起与否,图2(b)告诉我们非激活态最保守的特征其实是TM3与TM7之间的远离,而在激活态中,TM3与TM7的残基之间具有广泛的接触。因此,我们认为,TM7的运动尽管不如TM6剧烈,同样是GPCR激活所必需。
从序列角度看,A家族GPCR共同激活通路上的35个残基具有较强的序列保守性,与之对应的是配体结合区域和G蛋白耦合区域的残基组成非常多样。我们认为,GPCR共同激活通路中的残基构成一个负责激活的模块,解放出配体结合区域和G蛋白耦合区域来快速演化。这或许是GPCR可以使用一个简单的折叠模式来实现如此多样化功能的一个原因。
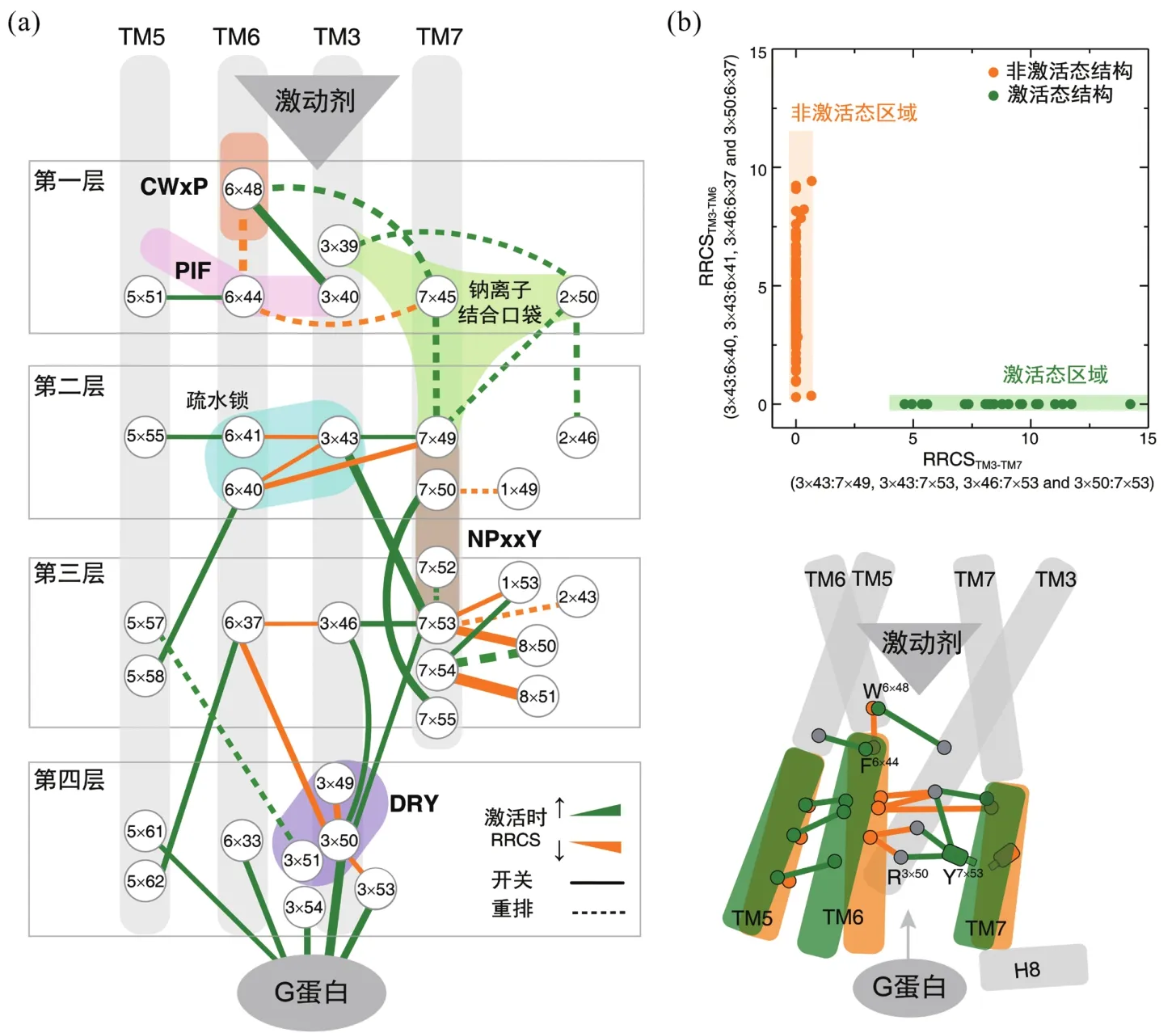
图2 A家族GPCR的共同激活机制[43]:(a)残基水平的共同激活通路,由在激活时具有保守构象变化的34组残基对构成;(b)激活时跨膜螺旋的构象变化
2.2 激活机制指导的组成性突变设计
GPCR的激活需要共同激活通路上各残基的参与,因此理论上可对这些关键残基进行干扰以实现激活信号的关闭或开启。为此,我们对共同激活通路上的残基进行理性突变设计以便将受体锁定激活构象或非激活构象,这样受体将处于组成性激活(不需要激动剂即可产生激活信号)或失活(即使有激动剂也不产生激活信号)。A2A腺苷受体是研究较多的A家族GPCR,它耦合激活型G蛋白(G),在结合激动剂后激活cAMP通路使得胞内cAMP含量增加。我们以A2A腺苷受体为例进行理性设计,最终证实15个设计的组成性激活突变中的6个、20个设计的失活突变中的15个都被cAMP功能实验证实,且它们的配体结合能力基本不受突变影响,这充分说明这些位点在GPCR激活过程中扮演着重要角色。此外,我们还对耦合抑制型G蛋白(Gi)的5-羟色胺受体1B(5-HT1B)和耦合激活型G蛋白的5-羟色胺受体7(5-HT7)进行基于GPCR共同激活通路的突变设计,实验证实这些突变体具有类似的组成性激活或失活效应。
2.3 激活机制与致病突变
GPCR的正常运作对于维持人体健康至关重要。GPCR的某些氨基酸突变可能会使GPCR功能紊乱并导致尿崩症、甲状腺功能亢进、性腺功能减退症、糖皮质激素缺乏症等遗传学疾病[44]。为了研究GPCR信号转导与疾病的关联,我们从数据库和文献上共收集人类GPCR的435个疾病相关突变,发现其中有25%的突变发生在GPCR的共同激活通路上,这一比例显著高于配体结合区域(20%)或G蛋白结合区域(7%)。从突变发生率看,致病突变明显富集在共同激活通路上,比配体结合区域高2.5倍,比G蛋白结合区域高3.5倍。实际上,GPCR的共同激活通路也为我们理解致病突变机制提供思路,如血管加压素V2受体(vasopressin V2 receptor,V2R)的I1303×43N突变会使得受体成为组成型激活,导致肾原性抗利尿激素分泌失调综合征(N-SIAD)[45]。I1303×43F则使受体失活,产生肾原性尿崩症(NDI)[46]。这可能是由于I130N/F会削弱/增加疏水锁的稳定性,削弱/增强TM3与TM6间的作用,使受体的TM6更易/难翘起,导致受体处于激活/失活状态,再经过下游信号通路的放大和转导,最终导致疾病的产生。
3 展望
GPCR激活机制是理解GPCR功能和理性药物设计的钥匙。伴随着过去20年GPCR结构解析的巨大发展,以及单分子、单残基甚至单原子分析技术的不断进步,我们对GPCR的认识日益深入,对配体激活受体、受体耦合G蛋白等关键问题也有了更丰富的认知,但距离全面理解GPCR的激活机制(家族共性和受体个性)仍有相当距离。从药物研发角度看,如何设计药物以精细调控GPCR的信号转导输出,如偏向性信号通路、亚型选择性、药物效率等重大问题,都绕不开GPCR的激活机制。总之,GPCR的激活机制研究内涵深刻,任重而道远,需要科研工作者们的不懈努力。
