六方氮化硼单层中一种(CN)3VB 缺陷的第一性原理计算*
2021-02-06孙志海黄强张颖黄鹏儒植慧茵邹勇进徐芬孙立贤
孙志海 黄强 张颖 黄鹏儒† 植慧茵邹勇进 徐芬 孙立贤‡
1) (桂林电子科技大学材料科学与工程学院,广西信息材料重点实验室,广西新能源材料结构与性能协同创新中心,桂林 541004)
2) (广西师范大学物理科学与技术学院,桂林 541004)
二维六方氮化硼(hBN)的点缺陷最近被发现可以实现室温下的单光子发射,而成为近年的研究热点.尽管其具有重要的基础和应用研究意义,hBN 中发光缺陷的原子结构起源仍然存在争议.本文采用基于密度泛函理论的第一性原理计算,研究hBN 单层中一种B 空位附近3 个N 原子被C 替代的缺陷(CN)3VB.在hBN的B 空位处,3 个N 原子各自带一个在平面内的悬挂键及相应的未配对电子,而通过C 替换可以消除未配对的电子.系统研究了(CN)3VB 缺陷的几何结构、电子结构以及光学性质,结果表明,缺陷可以由一个对称的亚稳态经过原子结构弛豫变成1 个非对称的、3 个C 原子连在一起的基态结构.缺陷的形成在hBN 中引入了一些由缺陷悬挂σ 键及重构的π 键贡献的局域缺陷态.这些缺陷态可以导致能量阈值在2.58 eV 附近的可见光内部跃迁.本文的工作有助于进一步理解hBN 中点缺陷的构成及光学性质,为实验上探讨发光点缺陷的原子结构起源及其性质提供理论依据.
1 引 言
固体中的点缺陷,如本征空位、杂质原子及其复合物等,是凝聚态物理研究的主要对象之一,也是现代量子技术应用具有潜力的基本单元,包括:量子传感[1−4]、量子纳米光子学[5−9]、量子信息处理[10,11]、量子通信[12−14]和计量学[15].其中具备单光子发射(single photon emission,SPE)是实现这些技术的一个重要前提.之前的研究主要集中在具有单光子发射的宽带隙半导体如金刚石、碳化硅等材料,虽然金刚石NV–色心是目前实现量子技术的首选平台,但是三维(3D)材料本身的局限性[16−18]以及最近在其他材料上发现的有趣色心,表明NV–并不是最佳的选择.二维(2D)材料正迅速成为基于光子的量子信息技术的新平台[19−28],与3D晶体相比,2D 材料的缺陷更容易读取和控制缺陷的性质和信息.因而,在2D 材料六方氮化硼(hBN)点缺陷处的单光子发射体(SPE)[29,30]的发现激发了人们对2D 晶体中光学点缺陷研究的热情.作为2D 材料,hBN 在常温常压下可以稳定存在,且hBN与半金属的石墨不同,hBN 中B—N 键的离子性质导致了约6 eV 的宽带隙,使其在间隙内产生具有基态和激发态的光学缺陷.hBN 的缺陷单光子发射器已经被证实具有一系列理想的特性,包括高发射率、室温稳定性、相对较强的零声子线以及易于与其他光学组件集成的特性[9,26,30−33].
实验上制备hBN 的过程中会产生本征空位和杂质元素的掺杂(如碳)[34−39],已有研究证明了碳杂质对hBN 材料的发光有很大影响.2012 年,Park等[40]证明与碳原子取代有关的杂质态及其相互作用决定了碳掺杂后hBN 的电子结构和性质,并且发现碳团簇中局部杂质态的堆叠在hBN 的宽间隙中形成了一组离散的能级.2018 年,Weston 等[41]研究了hBN 中天然缺陷和杂质的性质,排除了几种与缺陷相关的单光子发射可能性,并提出碳杂质及其复合物是可能的发光中心.2020 年,Wang等[42]利用不同浓度的碳掺杂hBN 提高基于hBN的深紫外光电探测器的性能.Mendelson 等[43]通过离子注入实验,确认只有碳注入hBN 才能在可见光谱范围内产生SPE,并提出带负电荷的B 空位附近单个N 原子被C 替代的缺陷结构(CNV−B)是能够解释实验中光谱的合适缺陷.Koperski 等[44]发现hBN 的碳富集可以在可见光和近红外光区域以共振形式激活材料的光学响应,并且在带隙中形成两个与碳相关的辐射中心.
尽管如此,对于2D hBN 发光点缺陷的研究仍然处于起步阶段,对于发光点缺陷的原子结构起源仍然存在很多争议.通过密度泛函理论(density functional theory,DFT)计算及分析已经为实验上观察到的发射光谱提供了一些候选的缺陷主体,但有关缺陷内部原子结构的很多结论还没有达成共识[41,43],对于hBN 中的点缺陷及其性质仍然需要进一步的研究.本文采用基于DFT 的第一性原理计算方法研究了hBN 单层中一种B 空位附近3 个N 原子被C 替代的缺陷(CN)3VB.通过原子结构、形成能、电荷态等的计算分析其热力学稳定性,通过能带结构、波函数等分析缺陷态在带隙中的位置及其原子轨道贡献,通过介电函数、吸收系数等分析其光学性质,并预测发光光子能量等.为深入理解hBN 中发光缺陷,并在实验上寻找单光子发射体提供了理论指导.
2 理论模型与计算方法
VASP (viennaab-initiosimulation package)是基于DFT 的第一性原理计算软件包[45−47],其可靠性已得到了大量研究的验证.本文通过DFT 采用Perdew-Burke-Ernzerhof (PBE)[48]的广义梯度近似描述电子之间的交换关联.采用投影缀加平面波[47]方法描述离子实与价电子的相互作用,赝势计算考虑的原子核外层电子组态分别为B 2s22p1,N 2s22p3,C 2s22p2.经过收敛测试,平面波截断动能Ecutoff= 500 eV,K点取6×6×1,使用自洽循环求解Kohn-Sham 方程的能量收敛值设为2 ×10–4eV/atom,最大应力控制在0.01 eV/Å以内.在计算空位点缺陷VB和碳掺杂缺陷(CN)3VB的同时,为了进行对比,计算了本征hBN 单层的情况.hBN 的原胞包含1 个N 原子和1 个B 原子.为了实现点缺陷和掺杂并且避免相邻超胞的相互影响,采用5×5 的超胞,即由25 个N 原子和 25 个B 原子组成的包括50 个原子的hBN 超胞.计算分为两部分,首先进行几何结构优化,然后在达到最稳定结构的基础上,对优化后体系的电子结构和光学性质进行高精度计算.
对于1 个带qα∆Hf(α,q)电荷的 缺陷,其形成能可以通过以下公式进行计算[49,50]:

其中

E(α,q) 是电荷态为q的缺陷α的总能量,E(host)是没有缺陷的宿主材料的总能量,EF是宿主材料价带的最大值(valence band maximum,VBM),µi是元素i的化学势,参考固体或气体元素的能量E(i) ,ni代表添加 (n>0)i类型元素或者减少(n<0)i类型元素的数量,q是电荷价态.当hBN单层处于平衡状态时,µB和µN应满足µB+µN=µBN,µB和µN的值由生长条件决定.当计算hBN单层中C 杂质的形成能时,µC从石墨计算而来.
热力学转变能级ε(q/q′) 被定义为费米能级位置,在该费米能级位置以下,带q电荷的缺陷是稳定的,在该费米能级位置以上,则带q′电荷的缺陷是稳定的,它可以通过以下公式计算得到[51]:

其中Ef(Dq;EF=0) 是费米能级为价带最大值(EF=0) 时,Dq的形成能,因此热力学转变能级ε(q/q′) 由费米能级位置确定,在该位置上,电荷态q和q′的形成能相等.
3 计算结果与讨论
3.1 本征hBN 单层
计算零温零压时,hBN 单层沿Brillouin 区高对称点方向的能带结构,选取费米能级EF附近–3—6 eV 的能带结构(其中EF= 0),hBN 单层价带的顶点在K点,而导带的底点在Γ点,即价带的最高点与导带最低点不在同一点,故hBN 单层属于间接带隙.这与Weston 等[41]计算的结果一致.hBN 单层的带隙宽度为4.366 eV,大于2.2 eV,因此可归为宽带隙半导体,实验中的hBN 的带隙约为6 eV[52,53],与本文计算结果的差异在27%,显然PBE 计算低估了h-BN 的带隙,这是由于DFT计算的是基态近似的结果,而在真实体系中的能隙属于激发态,这种情况通常会出现在宽带隙半导体中,但并不影响对hBN 单层电子结构的分析和研究.Huang 和Lee[50]通过Heyd-Scuseria-Ernzerhof(HSE)杂化泛函计算出纯净hBN 5.56 eV 的带隙,与实验结果更为接近.本文主要研究hBN 中碳掺杂缺陷的电子结构、光学性质等,虽然HSE 杂化泛函能够得到更接近实验值的带隙,但是目前并没有研究证明对于缺陷能级的计算,HSE 杂化泛函计算比PBE 计算更加准确,所以考虑到HSE 杂化泛函的计算量比较大,本文采用PBE 计算.
本征hBN 单层如图1 所示,其中图1(a)为本征hBN 单层的俯视图和侧视图; 图1(b)为模拟的hBN 单层表面扫描隧道显微镜(scanning tunneling microscope,STM)形貌,由于N 原子的电负性比B原子的电负性强,所以电荷主要集中于N 原子周围; 图1(c)是本征hBN 单层的能带投影图,可以看出,价带的边缘由N 原子的 pz轨道贡献,而导带的边缘主要由B 原子的 pz轨道贡献.
3.2 hBN 单层的硼空位缺陷
VB优化后的局部结构如图2(a)所示.对称的硼空位(VB)处三个最近的N—N 键长度均为2.647 Å,比优化前增加了0.135 Å.当打破VB的对称性后,非对称硼空位(asymmetry VB,as-VB)如图2(d)所示,自旋向上通道中的一个单一缺陷能级上升到间隙中,自旋向下通道的简并能级分裂为两个单一能级.as-VB自旋电荷密度图如图2(f)spin 所示,由原来的自旋向下变为自旋向上和自旋向下.在能量空间上,图2(d)中标记有1,2 的缺陷能级主要由N 原子的 py轨道贡献,标记有3 的缺陷能级主要由N 原子的 px轨道所贡献.在实空间上,Γ点处波函数图中标记有1,2,3 的缺陷能级均形成σ 键类型,这与能量空间上缺陷能级起源于空位处N 原子的 px和 py轨道一致.

图1 本征hBN 单层 (a) hBN 的俯视图和侧视图; (b) 模拟的hBN 单层表面STM 形貌,加载电压为–2 V,探针与原子表面的距离为0.479564 nm; (c) hBN 的能带投影图Fig.1.Intrinsic hBN monolayer: (a) Top view and side view of hBN; (b) the simulated hBN single-layer surface STM morphology,loading voltage is –2 V,the distance between probe and atomic surface is 0.479564 nm; (c) energy band projection view of hBN.
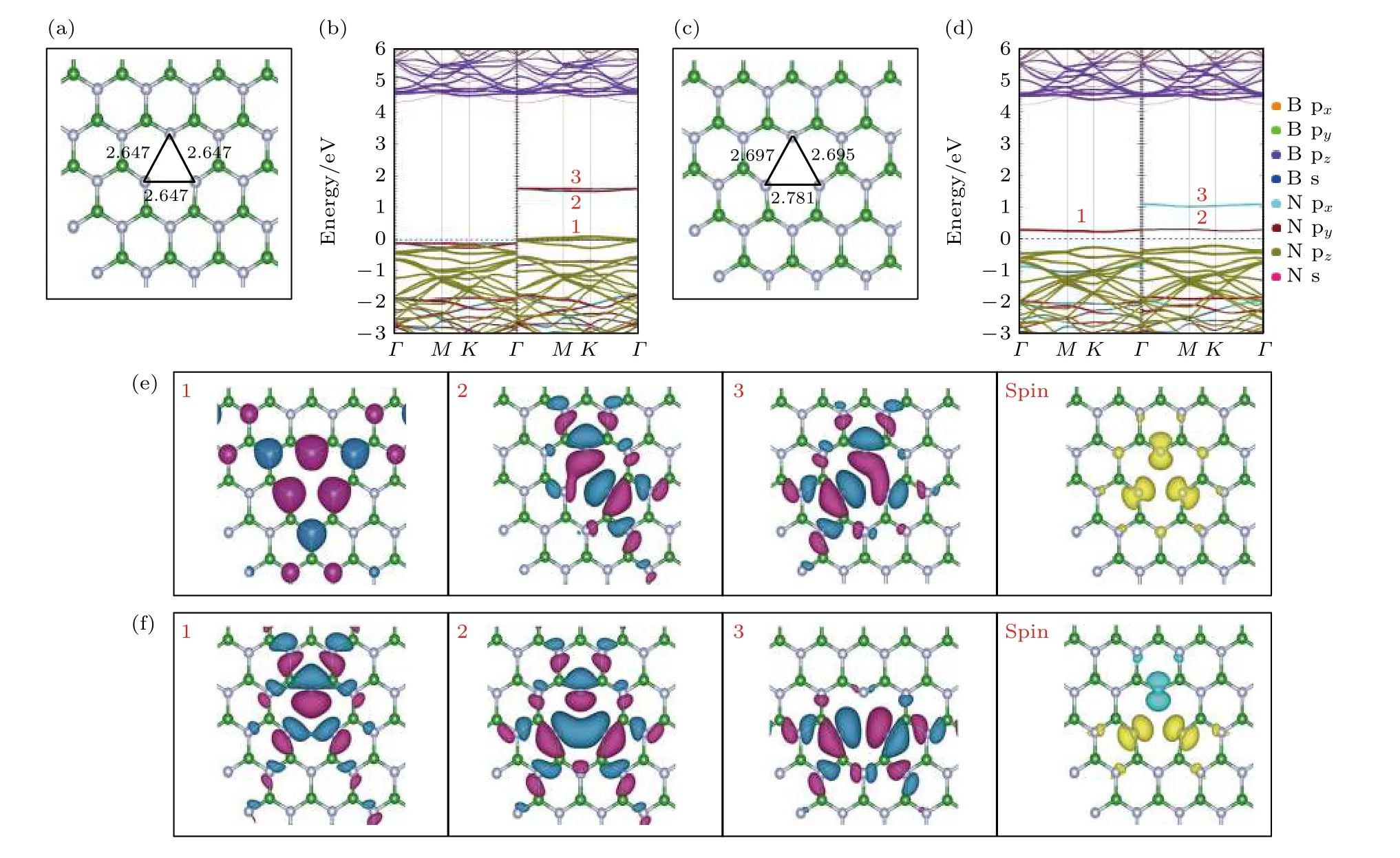
图2 VB 和as-VB 的电子结构 (a),(c)分别为VB 和as-VB 优化后的局部结构俯视图; (b),(d)分别为VB 和as-VB 的能带投影图;(e),(f)分别为VB 和as-VB 的缺陷能级在G 点处波函数的俯视图和自旋电荷密度图Fig.2.Electronic structure of VB and as-VB: (a),(c) The top views of the optimized partial structure of VB and as-VB,respectively;(b),(d) the energy band projection diagrams of VB and as-VB,respectively; (e),(f) the top view of the G-point wave functions of the defect levels and spin charge density of VB and as-VB,respectively.
在hBN 单层中的每个B 原子周围都有3 个N 原子.当除去一个硼原子形成VB时,会留下3 个N 2 sp2和3 个N 2 pz的悬空键.这些悬空键结合形成局部对称 ( aσ,aπ) 和较高价态的反对称(eσ,eπ)分子轨道.由前面单个硼原子空位的能带投影图分析得出,自旋通道中共有3 个未占据的缺陷态,每个缺陷态最多可以接受一个电子,所以VB可能存在0 至–3 的电荷态.图3(a)—(c)分别为 V−B1,V−B2,V−B3结构优化后的局部俯视图.随着电荷数的增加,库仑排斥力增强,N—N 键的长度增加,缺陷结构向外扩张.V−B1,V−B2,V−B3的能带投影图如图3(d)—(f)所示,当VB处于–3 价态时,由缺陷能级进入到导带中,所以 V−B3不能完全实现,VB的电荷态受近自由电子状态(nearly free electron,NFE)所影响[50].
通过(1)式和(2)式计算在富氮和贫氮条件下,V0B,V−B1,V−B2,V−B3的 形 成 能,如 图4 所 示.V0B在富氮和贫氮条件下的形成能分别为7.58 和10.11 eV.此外,还计算了富氮条件下VB的不同价态之间的热力学转变能级 ε (q/q′) ,其中 ε (0/−1) ,ε(−1/−2) ,ε (−2/−3) 分别为0.36,2.55,3.44 eV,我们的计算结果均与文献[50]接近.
3.3 hBN 单层碳掺杂的缺陷
由前面分析可知,硼空位缺陷附近的3 个氮原子(N 2s22p3)各自带一个在平面内的悬挂键及相应的未配对电子,而在元素周期表中,碳原子的价电子数比氮原子的价电子数少1 个,当用碳原子替换氮原子时,可以消除缺陷中未配对的电子,因此本文在VB的基础上,采用碳原子取代硼空位处的3 个氮原子,构成(CN)3VB的缺陷.(CN)3VB优化后的局部结构俯视图如图5(a)所示,图中C—C 的键长均为1.943 Å,这比未掺杂之前VB最近邻的N—N 键长缩短了将近26.6%.这主要是由于碳原子的电负性比氮原子的电负性弱,C 替换N 之后C-B 的成键能力比B—N 的成键能力弱,所以C—B 化学键较长,从而导致空位附近的碳原子向空位靠拢.

图3 VB 在不同价态的电子结构图 (a)−(c)分别为 , , 优化后的局部结构俯视图; (d)−(f)分别为 , ,V−B3的能带投影图Fig.3.Electronic structure diagrams of VB in different valence states: (a)−(c) The top views of the optimized partial structure diagrams of V −B 1 ,V −B 2 ,and V −B 3 ,respectively; (d)−(f) the energy band projection diagrams of V −B 1 ,V −B 2 ,and V −B3 ,respectively.
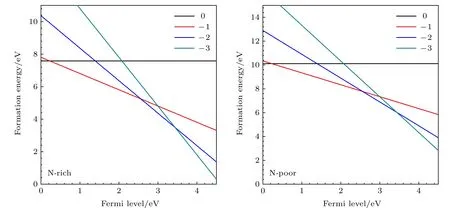
图4 在富氮和贫氮条件下,不同价态的VB 形成能为费米能级的函数Fig.4.Formation energies of VB with different valences as a function of Fermi level under the nitrogen-rich and nitrogen-poor conditions.
(CN)3VB的能带投影图见图5(b),自旋向上的通道中有2 个占据的缺陷态和2 个未占据的缺陷态,而自旋向下的通道中有4 个未占据的缺陷态,这 与 计 算 得 出 的 总 磁 矩 为 2 µB相 对 应.(CN)3VB的自旋电荷密度图如图5(e) spin 所示,(CN)3VB的磁矩主要是由硼原子空位处附近的3 个碳原子贡献.在能量空间上,(CN)3VB的能带投影图见图5(b),标记有1,2 的缺陷能级主要由C 原子的pz轨道贡献,标记有3,4 的缺陷能级主要由C 原子的 px和 py轨道贡献.在实空间上,在图5(e) G 点处的波函数图中标记有1,2 的缺陷能级是π 键类型,标记有3,4 的缺陷能级则是σ 键类型.
打破原先结构的对称性,构成非对称的碳掺杂结构(asymmetry (CN)3VB,as-(CN)3VB),如图5(c)所示,C3 与C1 和C2 相互成键,并且键长均为1.31 Å,相比于对称结构的键长缩短将近47%和45%,碳原子聚集,导致空位中缺陷能级的交换分裂,as-(CN)3VB的能带投影图见图5(d),原先由C原子的 pxy,pz轨道分别形成的双简并缺陷能级均分解为两个单一的缺陷能级.其中自旋向上(向下)通道各有一个单一的缺陷能级进入价带,自旋向上与自旋向下的缺陷能级的分布高度对称,总磁矩为0.在as-(CN)3VB能带投影图中标记有2 的缺陷能级由原来C 原子的 pz轨道变为C 原子的px和 py轨道贡献,形成σ 键类型,而标记有3,4 的缺陷能级由原来C 原子的 px和 py轨道变为B 原子的pz轨道贡献,形成π 键类型.

图 5 (CN)3VB 和as-(CN)3VB 的 电 子 结 构 (a),(c)分 别 为(CN)3VB,as-(CN)3VB 优 化 后 局 部 结 构 的 俯 视 图; (b),(d)分 别 为(CN)3VB,as-(CN)3VB 的 能 带 投 影 图; (e) (CN)3VB 缺 陷 能 级 在G 点 处 的 波 函 数 图 和 自 旋 电 荷 密 度 图; (f) as-(CN)3VB 缺 陷 能 级 在G 点处的波函数图Fig.5.Electronic structure of (CN)3VB and as-(CN)3VB: (a),(c) The top views of the optimized partial structure of (CN)3VB and as-(CN)3VB; (b),(d) the energy band projection diagrams of (CN)3VB and as-(CN)3VB; (e) the top view of the G-point wave functions of the defect levels and spin charge density of (CN)3VB; (f) the top view of the G-point wave functions of the defect levels of as-(CN)3VB.
由于在(CN)3VB和as-(CN)3VB的体系中均存在2 个占据的缺陷态和6 个未占据的缺陷态,因此可以预测(CN)3VB可能存在的电荷状态可以在+2 至–6 的范围内.优化后的局部缺陷俯视图分别如图6(a)—(d)所示.当(CN)3VB从–1 到–2 价态变化时,电子数增加导致碳原子之间库仑排斥力增大,C—C 键长由2.059 Å增加到2.154 Å.当(CN)3VB从+1 到+2 价 态 变 化 时,C—C 键 长 由1.825 Å缩短至1.712 Å,这是因为电子数减少,碳原子之间相互吸引力更强,导致缺陷结构进一步向内收缩.的能带投影图如图6(e)—(h)所示.对于(CN)3V−B1,费米能级上移,在自旋向下通道中,仅有一个C 原子 pz轨道的缺陷能级未被占用,产生了1µB的总磁矩.当向(CN)3V−B1中添加一个电子形成时,由于占据的电子之间的库仑排斥,缺陷能级向下移动进入价带,C 原子 px和 py轨道贡献的缺陷能级与导带混合,如果继续添加的电子可能被导带占据,从而无法确定添加的电子是否被缺陷态所占据,所以不能实现.在中,费米能级下移,并且仅在自旋向上通道中有一个C 原子π 键类型的占据态,说明添加的空穴到了缺陷能级.当(CN)3VB处于+2 价态时,自旋通道中没有占据的缺陷态,因此,(CN)3VB最多只能存在+2 价态.优化后局部结构的俯视图如图7(a)—(d)所示.非对称碳掺杂缺陷处于不同价态时,碳原子的聚集度均高于对称碳掺杂缺陷.as-(CN)3的能带投影图如图7(e) —(h)所示.从图7 可知,as-(CN)3VB同样存在从 –2 到 +2 的电荷价态.图8和图9 分别为(CN)3VB和as-(CN)3VB在富氮和贫氮条件下不同电荷状态的形成能.在富氮条件下,(CN)3V0B和as-(CN)3V0B的形成能分别为11.66和7.94 eV,as-(CN)3VB的形成能比(CN)3VB低了3.72 eV,这说明当碳掺杂hBN 时,更倾向于形成非对称的基态结构.本文还计算了(CN)3VB的不同价态之间的热力学转变能级 ε (q/q′) ,其中,ε(+2/+1) ,ε (+1/0) ,ε (0/−1) ,以及 ε (−1/−2) 分别为0.20,0.54,1.64 以及1.93 eV.而as-(CN)3VB的热力学 转 变能 级ε(+1/0) ,ε (0/−1) 和 ε (−1/−2) 分别为0.64,3.32 和3.96 eV,对比可知,非对称碳掺杂的热力学转变能级均高于对称的缺陷.

3.4 光学性质

图9 在富氮和贫氮条件下,不同价态的as-(CN)3VB 形成能为费米能级的函数Fig.9.Formation energies of as-(CN)3VB with different valences as a function of Fermi level under the nitrogen-rich and nitrogenpoor conditions.
复介电函数能够用来描述系统对电磁辐射的线性响应,这与光在介质中传播时,光子的电场扰动使介质中的电子发生跃迁有关,它是能带结构的宏观表象.复介电函数的虚部能从占据态和未占据态波函数之间的动量矩阵元素中计算出来,其实部能够通过Kramers-Kronig 方程从虚部中推导出来.复介电函数:

其中w表示入射光的频率,ε1(w) 表示复介电函数的实部,iε2(w) 表示复介电函数的虚部,i 表示虚数单位.本征hBN,VB,as-VB,(CN)3VB,as-(CN)3VB的复介电函数实部和虚部随入射光能量Ein的变化分别如图(10)(a)—(e)所示.在对称缺陷中,复介电函数实部和虚部分别在X和Y方向的张量相同,表现出各向同性,但在非对称缺陷中,实部和虚部分别在X和Y方向的张量不同,表现出明显的各向异性.当入射光能量Ein= 0 时,hBN,VB,as-VB,(CN)3VB,as-(CN)3VB的 介 电常数 值ε0(单位: C2·N–1·m–2)分 别 是2.43,33.35,3.50,32.47,3.22,因此对称的VB和(CN)3VB的屏蔽性都比较强.在低能区域(Ein< 5 eV),具有缺陷的hBN 在复介电函数的实部和虚部均出现了峰值,并且吸收边界出现红移,说明硼空位和碳掺杂缺陷提高了hBN 对可见光的光学响应程度.

图 10 0 K 和0 GPa 时hBN,VB,as-VB,(CN)3VB,as-(CN)3VB 的复介电函数Fig.10.Complex dielectric functions of hBN,VB,as-VB,(CN)3VB,as-(CN)3VB at 0 K and 0 GPa.

图11 0 K 和0 GPa 时hBN,VB,as-VB,(CN)3VB,as-(CN)3VB 的吸收系数Fig.11.Absorption coefficients of hBN,VB,as-VB,(CN)3VB,as-(CN)3VB at 0 K and 0 GPa.
吸收系数 I 和复介电函数的虚部iε2(w) 密切相关,因此可以通过介电函数的虚部得到光学吸收谱,如图11 所示,为了清楚显示吸收光谱在低能区间的吸收峰,图11(a)—(e)中的左图纵坐标较右图放大约6 倍,本征hBN 在可见光波段范围几乎不吸收光子,直到Ein= 4.4 eV 时,开始吸收光能量,这与前面计算出的本征hBN 的带隙为4.37 eV相符合.在硼原子空位与碳原子掺杂之后,吸收边界出现较大的红移,并且在0.2 eV 均出现了吸收峰,这可能是由于VB和(CN)3VB的价带穿越费米能级EF导致的.当打破对称结构时,as-VB和as-(CN)3VB的吸收峰发生了蓝移,且由吸收橙红色光转变为吸收青蓝色光为主.在较高能量的波段中,本征hBN 与有缺陷的hBN 的吸收光谱大致相同.
损失函数L 描述的是光电子在均匀介质中穿过时的能量损失情况,本征hBN 和缺陷hBN 的吸收函数如图12 所示,本征hBN 和缺陷hBN 在Ein=7 eV 和Ein= 18 eV 处具有明显的能量损失峰.但是,有缺陷的hBN 在低能区域(Ein< 5 eV)引入了许多损失峰,这与图11 吸收光谱在低能区时具有吸收峰相对应,并且经过碳掺杂后,hBN 的损失峰主要集中在可见光波段.说明具有空位和碳掺杂缺陷的hBN 在低能段时,能级间相互作用比较强.
为了探究hBN 发光缺陷的原子结构起源,根据前面计算的hBN 缺陷的电子结构和光学性质,对缺陷带隙中可能存在的内部能级跃迁(基态到激发态)进行分析.VB的电子结构模型如图13(b)所示,在价带以上0.41 eV 的潜在基态到价带以上2.07 eV 的潜在激发态之间有单一的跃迁,导致约1.7 eV 的跃迁,这与图13(d)中VB的介电函数虚部在Ein为1.7 eV 处的峰值相对应,在吸收光谱上对应波长为730 nm 的红色光.as-VB的电子结构模型如图13(e)所示,自旋向上和自旋向下通道中均存在价带以上0.14 eV 的潜在基态到价带以上0.89 eV 的潜在激发态之间的内部跃迁,而且两个自旋通道的内部跃迁几乎为简并能级,导致约0.75 eV 的跃迁,这与图13(f)中as-VB的介电函数虚部在Ein为0.75 eV 处的峰值相一致,为深红外光.
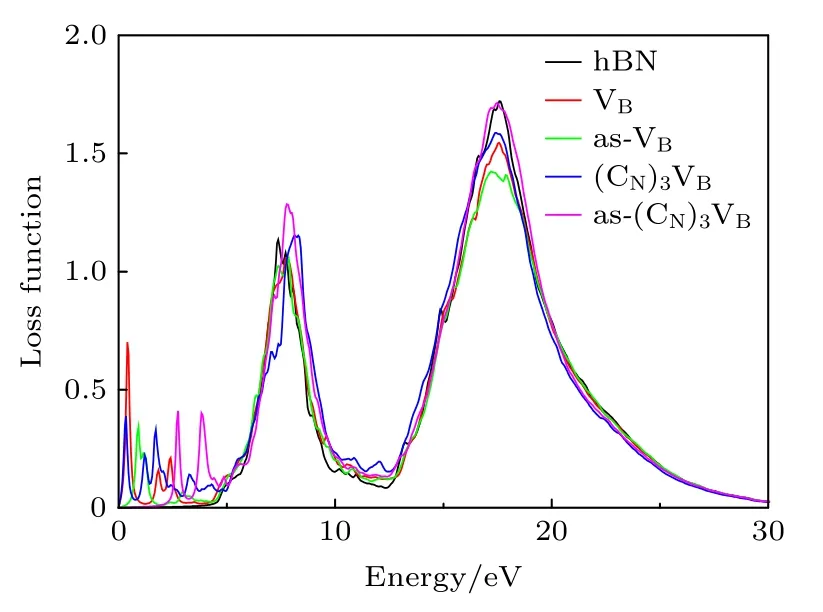
图 12 0 K 和0 GPa 时hBN,VB,as-VB,(CN)3VB,as-(CN)3VB 的损失函数Fig.12.Loss function of hBN,VB,as-VB,(CN)3VB,as-(CN)3VB at 0 K and 0 GPa.
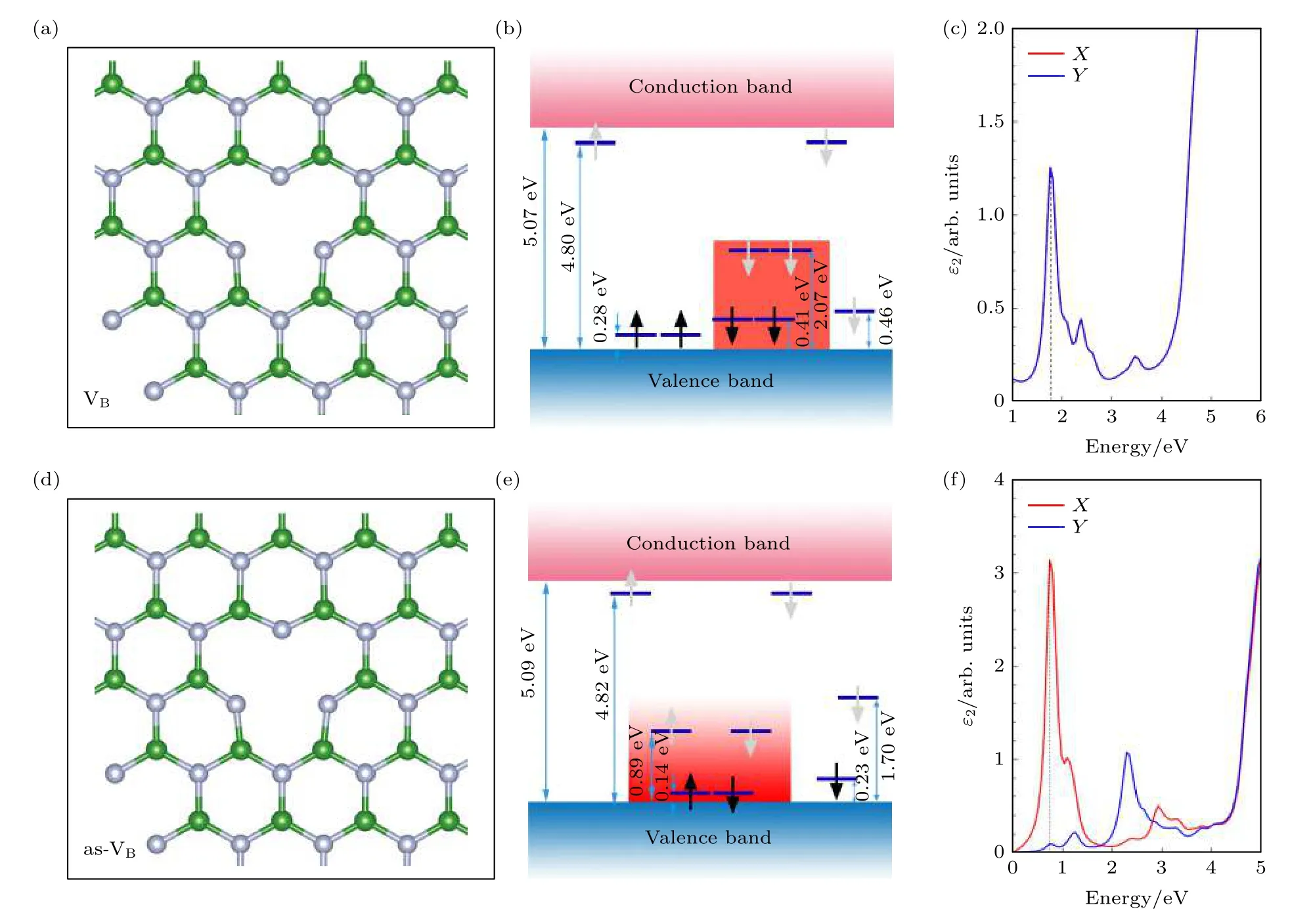
图13 hBN 单层硼原子空位的模型图 (a),(d)分别为VB 和as-VB 优化后的局部结构图; (b),(e)分别为VB 和as-VB 的电子结构模拟图,黑色箭头和灰色箭头分别代表占据态和未占据态; (c),(f)分别为VB 和as-VB 的介电虚部在X 和Y 方向张量Fig.13.Model diagram of boron vacancies in hBN monolayer: (a),(d) The optimized local structure diagrams of VB and as-VB;(b),(e) simulated electronic structures diagrams of VB and as-VB,where black and grey arrows indicate occupied and unoccupied states; (c),(f) the tensors of the dielectric imaginary part of VB and as-VB in the X and Y directions.
(CN)3VB的电子结构模型如图14(b)所示: 在自旋向上通道中,存在一个价带以上0.89 eV 的潜在基态到价带以上4.14 eV 的潜在激发态之间的单一跃迁,导致约3.25 eV 的光学跃迁; 在自旋向下通道中,存在一个价带以上0.46 eV 的潜在基态到价带以上4.51 eV 的潜在激发态之间的单一跃迁,导致4.05 eV 的光学跃迁; 分别与图14(c)中(CN)3VB的介电函数虚部在Ein约为3.25 和4.05 eV 处的峰值相对应,为波长307 和382 nm的紫外光.在非对称的碳掺杂缺陷中,as-(CN)3VB的电子结构模型如图14(e)所示,自旋向上和自旋向下通道中均存在价带以上1.30 eV 的潜在基态到价带以上3.88 eV 的潜在激发态之间的内部跃迁,两个自旋通道的内部跃迁能级为双简并能级,导致约2.58 eV的跃迁,与图14(f)中as-(CN)3VB的介电函数虚部在Ein为2.58 eV 处的峰值相一致,且其吸收光为波长570 nm 的青色光,这是与图11 中非对称碳掺杂缺陷的吸收光谱相符合,说明非对称的碳掺杂缺陷在可见光波段为单光子发射提供了一个潜在的跃迁途径.
4 结 论
本文在hBN 单层中构造了一种B 空位附近3 个N 原子被C 替代的缺陷(CN)3VB,并利用第一性原理系统地研究缺陷的几何结构、电子结构以及光学性质.通过计算发现碳掺杂之后,缺陷可以由一个对称的亚稳态经过原子结构弛豫变成1 个非对称的、3 个C 原子连在一起的基态结构.电子结构上,从对称到非对称缺陷结构的转换中,缺陷能级发生交互分裂,在hBN 中引入了一些由缺陷悬挂σ 键及重构的π 键贡献的局域缺陷态.在光学性质上,空位和碳掺杂缺陷明显提高了hBN 对可见光的吸收强度,并且都存在内部光学跃迁的可能性,还发现非对称碳掺杂缺陷可以导致能量阈值在2.58 eV 附近的可见光内部跃迁.本文的工作有助于进一步理解hBN中点缺陷的构成及光学性质,为实验上探讨发光点缺陷的原子结构起源及其性质提供理论依据.

图14 hBN单层碳掺杂的模型图(a),(d)(CN)3VB和as-(CN)3VB优化后的局部结构图;(b),(e)(CN)3VB和as-(CN)3VB的电子结构模拟图,黑色箭头和灰色箭头分别代表占据态和未占据态;(c),(f)(CN)3VB和as-(CN)3VB的介电虚部在X 和Y 方向张量Fig.14.Model diagram of carbon doping in hBN monolayer:(a),(d)The optim ized local structure diagrams of(CN)3VB and as-(CN)3VB;(b),(e)simulated electronic structures diagram s of (CN)3VB and as-(CN)3VB,where black and grey arrows indicate occupied and unoccupied states;(c),(f)the tensors of the dielectric imaginary part of(CN)3VB and as-(CN)3VB in the X and Y directions.
