丙烯氢氧环氧化动力学与反应器概念设计研究进展
2021-01-30杜威张志华段学志周兴贵
杜威,张志华,段学志,周兴贵
(华东理工大学化学工程联合国家重点实验室,上海200237)
引 言
环氧丙烷(PO)是仅次于聚丙烯的第二大丙烯衍生物,主要(约78%)用于合成聚醚多元醇[1]。近年来,随着聚氨酯产业的飞速发展,我国PO 消费量迅速增加,PO 产能随之急剧扩张[2]。截至2019 年,建成和在建的产能合计约870 万吨[3]。目前,PO 的工业生产工艺有氯醇法、共氧化法和过氧化氢液相环氧化法[4-7]。传统的氯醇法使用氯气为原料,对装置材质要求高,且产生大量废水废渣,已被限制发展,改良的氯醇法避免了废水废渣问题,但需配套氯碱装置,耗电量很大;乙苯/异丁烷共氧化法(PO/SM 和PO/TBA)流程较复杂,投资大,其中PO/SM 联产2.2~2.5 倍苯乙烯,PO/TBA 联产2.3 倍叔丁醇,其经济效益受联产物市场和价格的影响很大;异丙苯共氧化法(CHP)虽然避免了联产大量共生产物,但流程长,需要增加二甲基苄醇脱水和加氢反应装置,而且异丙苯氧化和异丙苯过氧化氢分解时伴有很多副反应,产生的大量副产物需要充分分离,因此物耗和能耗均较高;双氧水氧化法(HPPO)工艺简单、反应条件温和,但须配套高纯双氧水生产装置,增加了该工艺的成本,同时为了维持较高的PO选择性,反应在低沸点甲醇溶剂中进行,并在低PO浓度下操作,对PO 进行浓缩和分离时需要消耗大量的蒸汽,能耗因此居高不下[8]。
使用丙烯与H2、O2直接环氧化制PO 的方法被称为丙烯氢氧环氧化法(HOPO),该方法克服了HPPO的缺点[7],如图1所示,在投资和能耗方面具有其他PO 生产工艺不可比拟的优势,被称为“梦反应”[9-11],具有以下特点:(1)从原料到产物只需经过一步反应,工艺简单,原料价廉易得;(2)PO 选择性高,原子经济性好,CO2选择性通常低于5%;(3)反应在气相进行,未转化的丙烯、氢气和氧气沸点很低,易于与产物PO 和丙酮、丙醛等副产物分离;(4)反应可在较高温度(150~200℃)下进行,用沸腾水移热,产生的蒸汽可用于产品的分离和精制。Haruta等[12]在1998 年首次提出了使用Au/TiO2催化剂催化HOPO反应,他认为HOPO反应需要利用Au-Ti双功能催化剂[13-16]。Au 位催化H2与O2生成氢过氧化物种,之后转移至四配位的Ti 位上形成Ti-OOH 活性物种,Ti-OOH 再与C3H6发生环氧化反应生成PO[17-20]。但此时催化剂反应活性较低[21]。2012 年,Delgass 等[22]以钛硅分子筛TS-1 为载体首次制备出活性高且重复性好的Au/TS-1 催化剂,这一工艺开始真正引起工业界和学术界的重视。目前,国内外对于该工艺的开发仍处于催化剂的实验室研究阶段,Au/TS-1 催 化 剂 初 始PO 活 性(initial PO formation rate)较高,与早期乙烯环氧化制环氧乙烷的活性相当,可基本满足工业生产对催化剂时空产率的要求,但催化剂活性下降很快[18,22-24]。Zhou 等[25]通过研究发现,Au/TS-1 催化剂快速失活的主要原因是碳物种堵塞TS-1的微孔,使催化剂活性位数量减少,据此他们采用分子筛微孔被模板剂堵塞的未焙烧TS-1(即TS-1-B)作为载体选择性地将Au 纳米颗粒沉积在TS-1-B 的外表面,将催化剂的稳定性显著提高至30 h左右,PO的稳定生成速率也远高于之前文献报道的催化剂[23-24,26],近年Zhou等[27]合成出未焙烧的纳米TS-2-B 载体,采用沉积沉淀法负载Au 制备出Au/TS-2-B 催化剂,其选择性与Au/TS-1-B相当,但氢效(氢气的利用率)和稳定性得到显著提升,能够稳定运行100 h 以上,具有一定的工业化前景。
本文基于目前已报道的Au-Ti 催化剂,综述了近年来Au-Ti双功能催化丙烯氢氧环氧化的反应动力学,针对本反应中混合气体中可能存在的安全风险,总结了HOPO反应的反应器概念设计进展,最后展望了丙烯氢氧环氧化工艺可能的研究方向及工业化前景。
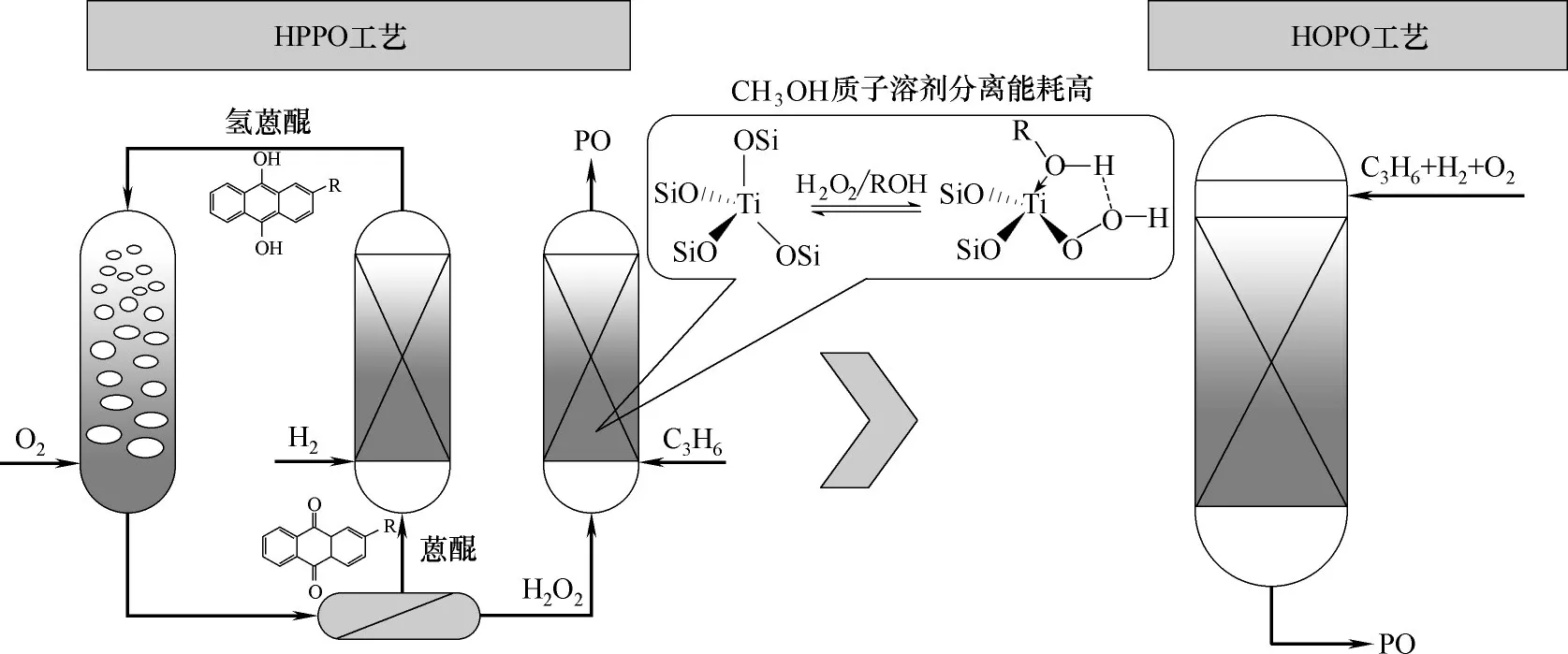
图1 HPPO与HOPO工艺比较Fig.1 Schematic diagram of HPPO&HOPO technology
1 Au-Ti 催化剂上的HOPO 反应动力学
1.1 主反应动力学
1998 年Haruta 等[12]首次报道Au/TiO2催化剂具有催化HOPO 反应的活性,然而Au/TiO2催化剂在HOPO 反应中存在活性低,且面临快速失活的问题,限制了其进一步应用。为深入理解反应与失活机理,Zhou 等[28]研究了Au/TiO2催化剂上的反应及失活动力学,并通过改变催化剂上Au的负载量进行动力学实验,使用实验点与预测值进行对比,验证了模型的可靠性。该模型假设整个反应的决速步为Au和TiO2载体界面边缘位点上过氧物种的形成,并假设反应气体均已经达到吸附平衡,采用E-R 机理,如图2 所示。结合图2(a)所示反应步骤,并基于一维非等温平推流反应器模型建立了动力学方程,其中反应①~④分别表示了反应器中氢气与氧气的消耗、产物PO 的生成、产物PO 的吸/脱附,以及PO 深度反应生成丙氧基物种(D)覆盖Ti 活性位点导致催化剂失活。
使用不同进料浓度的实验结果对上述方程进行联立求解,并建立了不同反应温度与催化剂负载量下包含催化剂失活的动力学模型。方程求解结果发现,除失活步骤外,其他反应步骤中反应速率常数ki与活化能Ea均符合Arrhenius 方程[29],进一步对比反应器出口PO 浓度的预测值与实验值,发现有较好的一致性。而失活步骤方程并不能很好地描述实验结果,由此他们认为PO 的深度反应导致失活的过程并非如假设所示为一步的反应过程,需要建立更详细的模型来描述。
Nijhuis 等[30-31]研究了在Au/TiO2催化剂上HOPO的反应动力学,结合原位红外光谱分析,提出了以双配位丙氧基物种为中间体的反应机理。首先通过红外光谱观察到丙烯在Au/TiO2催化剂上发生了不可逆的吸附,形成了双配位丙氧基物种,氢气和氧气同时存在会促使该物种的脱附,而在TiO2载体上仅观察到丙烯发生可逆的物理吸附[32]。因此他们认为丙烯吸附于Au 表面并在Au 表面发生活化,然后溢流到TiO2载体上,与Ti 位点的晶格氧生成吸附的双配位丙氧基物种。该物种被H2和O2形成的过氧物种协助脱附,生成PO,而催化剂失活主要是由于双配位丙氧基物种的深度氧化[33]。后来Nijhuis等[34]又结合X 射线近边吸收光谱(XANES)和扩展X射线吸收精细结构谱(EXAFS)表征进一步确定了丙烯吸附在Au位点上,并发现丙烯的吸附为π键的作用。他们还通过稳态同位素瞬态动力学分析(SSITKA)实验[35]发现吸附的产物中的氧与反应气中的氧数量总和要远大于对应的产物PO 生成速率,因此进一步确认了晶格O 原子在反应中发挥的作用。基于上述反应机理及平推流反应器模型建立了反应动力学,该模型中假设Au/TiO2催化剂上H2/O2/C3H6发生HOPO 反应的气体转化率很低,因此在模型中将其浓度作为定值处理,只考虑了反应中PO浓度的变化。

图2 Au/TiO2催化剂上丙烯氢氧环氧化反应动力学(采用E-R机理)[28]Fig.2 The kinetics of HOPO reaction over Au/TiO2 catalyst(based on E-R mechanism)[28]
如图3 所示,动力学方程中,分别描述了PO 轴向浓度随时间的变化、Au 位/Ti 位附近的Ti 位点吸附的PO 浓度随时间变化、催化剂的失活以及再生。其中失活步骤基于反应中PO 深度氧化形成羧酸盐/碳酸盐物种,失活速率方程被认为是对吸附PO 浓度的一级反应。但该动力学未考虑更复杂的情况:如水存在的竞争吸附,以及副产物的生成路径。
随后在较低反应温度范围(≤365 K,此时反应的选择性很高),分别考察了反应温度和进料流率变化对反应结果的影响,经过实验值与模型值的比较发现该方程组能较好地描述出实际情况下流速和温度变化时该反应的反应器出口PO 浓度变化[30],如图4所示。
Nijhuis 等[36-37]使用微反应器研究了Au/Ti-SiO2催化剂上宽浓度范围下(包含爆炸范围)的反应及失活动力学,提出Au颗粒上活性过氧物种的形成是反应决速步骤,并假设PO 生成速率rPO正比于初始速率rPO,0与催化剂上活性位含量:

由于催化剂的失活来源于PO 的深度反应,因此假设失活速率正比于PO 生成速率与活性位含量:

图3 Au/TiO2催化剂上丙烯氢氧环氧化反应的动力学(双配位丙氧基物种为中间体)[30]Fig.3 The kinetics of HOPO reaction over Au/TiO2 catalyst(double coordinated propoxy species as intermediate)[30]
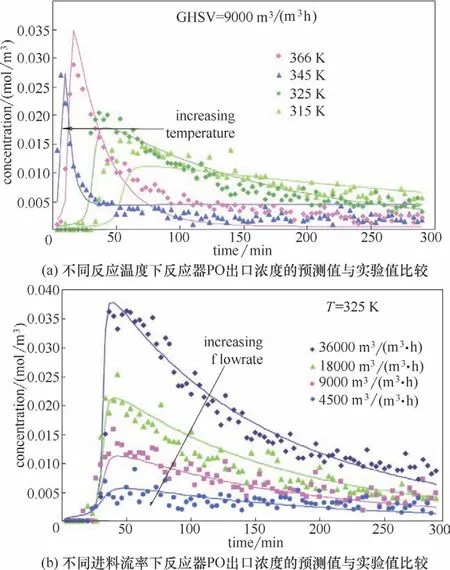
图4 Au/TiO2催化剂上丙烯氢氧环氧化反应的动力学预测值与实验值比较Fig.4 Comparison between measured and modeled kinetics of Au/TiO2 catalyst for HOPO reaction
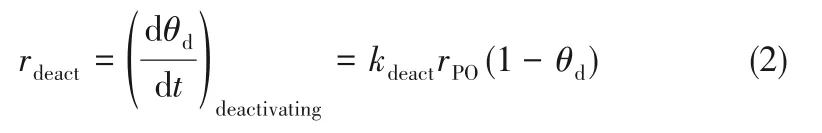
再生速率正比于失活位点的含量:
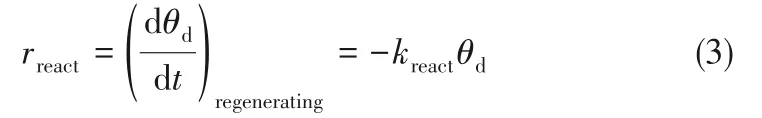
且失活位点的生成速率存在如下平衡:

联立求解后得出动力学方程:
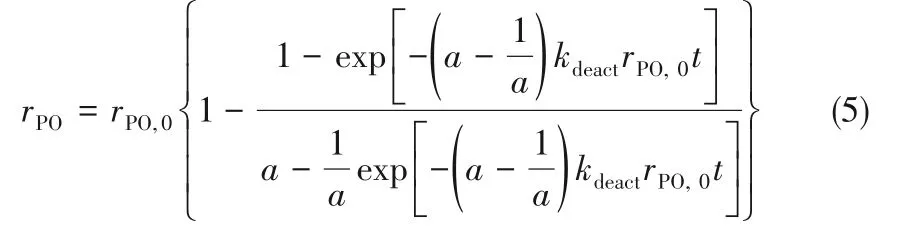
该动力学研究结果表明,PO 生成速率受H2浓度影响最大,增加H2浓度可以显著提高PO 的生成速率,但提高氢气浓度会增加过氧化氢物种的生成,从而加速催化剂的失活;失活主要由于PO 的深度氧化,提高丙烯浓度时,过氧化物中间体浓度会因环氧化更多的丙烯而减少,生成更多的PO,从而减缓失活过程。增加O2浓度可降低催化剂失活的速率,并且可加速失活催化剂的再生,如图5 所示。在HOPO 反应中选择适中的氢气浓度、较高的丙烯浓度和氧气浓度进行反应,有利于得到最佳的催化性能。

图5 Au/Ti-SiO2催化剂上动力学结果[37]Fig.5 The kinetics results of HOPO reaction over Au/Ti-SiO2 catalyst[37]
Delgass 等[38]研究了Au/TS-1 催化剂上的HOPO反应动力学,其首先采用理论计算研究了金原子上丙烯在H2/O2共存条件下直接气相环氧化反应机理[39-40]。该机理主要由以下几个步骤组成:(1)O2分子吸附在金团簇上形成Au-O 键;(2)H2分子在吸附的O2协助下发生解离吸附生成OOH 物种;(3)Au-OOH 物种上亲电性的O 原子(靠近Au 的O 原子)进攻C3H6上的碳碳双键形成PO。该反应机理被称为协同机理(cooperative mechanism)。DFT 计算得出生成PO 的活化能为19.6 kcal/mol,与之前文献报道的涉及Ti 催化烯烃氧化反应的活化能很接近(15~20 kcal/mol),且计算发现生成水的反应速率常数远高于丙烯环氧化反应的速率常数,这预示着丙烯环氧化反应是整个反应的决速步骤[40-41]。DFT 计算还发现Au/TS-1 催化剂中邻近纳米金团簇(Au3)的Ti缺陷位点上生成Ti-OOH 的活化能为32.1 kcal/mol,比决速步丙烯环氧化反应(Eact=20.3 kcal/mol)和不含Au 的Ti 缺陷位上生成Ti-OOH 的活化能(16.8 kcal/mol)都要高,这预示着在Au/TS-1 催化剂上生成Ti-OOH 物种在动力学上是受限的,环氧丙烷更有可能是通过C3H6与H-Au-OOH 物种直接反应生成。
基于上述机理推导得出Au/TS-1催化剂上环氧丙烷生成速率表达式[式(6)],存在如下简化:K[A]/(1+K[A])≈(K[A])a,因此式(6)可化简为如式(7)所示的幂函数型速率表达式。通过Au/TS-1催化剂上所得动力学实验结果进行拟合,发现C3H6、H2、O2对PO生成的反应级数分别为:0.18 ± 0.04,0.60 ± 0.03,0.31±0.04。若假设该幂函数型速率表达式中氧气的级数2m=0.28,则据此可得氢气的级数为0.64,与上述实验拟合结果相一致。由协同机理推导得出的氢气与氧气反应级数之间的关系可合理解释由动力学实验数据拟合所得氢气与氧气的反应级数,证明了上述机理与动力学模型的合理性。

Oyama 等[42]研 究 了Au/Ti-TUD 催 化 剂 上 的HOPO 反应动力学。他们提出H2、O2、C3H6对PO 生成速率的反应级数[43]分别为0.54、0.24、0.36,认为反应由Au 与载体上Ti-OH 两个位点上的独立反应循环组成,分别为Au 位点上过氧化物的生成反应与Ti-OH上丙烯环氧化生成PO的反应,且环氧化速率要远远大于过氧化物生成速率,由此认为HOPO 反应更可能遵循两个反应先后发生的机理(次序机理),而非同时发生的机理(协同机理),即过氧化物先在Au 位点生成,再迁移到Ti 位点与吸附的C3H6发生环氧化反应生成PO。通过对比Au/Si-TUD、Au 以及Au/Ti-TUD 上反应气体的吸附量,发现含Ti 载体的气体吸附量远大于无Ti 载体的,因此他们认为多数吸附发生于Ti 位点,更有可能是Au 颗粒附近的Ti 位点。根据不同假设,进一步推导获得了六种基本动力学方程:(1)H2/O2/C3H6竞争吸附的单位点模型;(2)H2/O2/C3H6竞争吸附的双位点模型;(3)H2/O2/C3H6独立吸附的三位点模型;(4)H2/O2/C3H6独立吸附且氢气解离吸附的三位点模型;(5)PRL 幂 函 数 级 数 模 型;(6)非 均 匀 表 面 的Frumkin-Temkin (F-T) 模型。以及由三种复杂机理推导方程:(1)次序机理的L-H 模型;(2)协同机理的L-H 模型;(3)由次序机理L-H 模型简化得到的L-H/PRL 复合模型,如表1 所示。通过F 检验、模型相对概率计算等统计学方法检验了上述模型对实验数据拟合及预测的合理性,得出统计意义上最好的模型是由次序机理简化得到的L-H/PRL复合模型。H2/O2/C3H6-TPD 结果显示,气体吸附量次序为O2>C3H6>H2,与由单位点、双位点、三位点吸附以及H2解离吸附的L-H 机理模型、非均匀表面的F-T 模型以及PRL 模型的平衡吸附常数均能够吻合,但上述模型对于实验结果的拟合效果并不佳,而由协同机理简化而来的L-H/PRL 复合模型级数关系为H2级数>C3H6级数>O2级数,与TPD实验结果中气体的吸附量次序一致,进一步说明了该模型的可靠性。由于本研究中丙烯转化率很低,仅为1.4%,因此并未考虑PO 的共吸附对动力学模型的影响。
他们还提出Au/Ti-TUD 催化剂上HOPO 反应[43]生成PO 的活化能为43 kJ/mol,小于生成H2O(51 kJ/mol)和CO2的活化能(80 kJ/mol)。这与Zhou 等[44]在Au/TS-1-B 催化剂上得到的活化能规律类似,他们发现6 种产物生成的活化能(kJ/mol)次序为CO2(92.2)>丙醛(85.6)>丙酮(73.7)>丙烯醛(71.1)>乙醛(60.5)>PO(29.2)。Oyama 等[43]认为H2O 主要是由H2和O2的反应形成的,而CO2是PO 进一步氧化的产物。

表1 Au/Ti-TUD催化剂上不同动力学方程拟合结果[42]Table 1 Summary of kinetics fits of HOPO reaction over Au/Ti-TUD catalyst[42]
1.2 考虑产物抑制效应的反应动力学
HOPO 反应产物PO 对于主反应表现出一定的抑制效应。Delgass 等[45]采用无梯度反应器(CSTR)研究了Au/TS-1 催化剂上HOPO 反应动力学。实验结果显示,当在原料气中通入PO 后,PO 生成速率明显降低,这表明产物PO 在活性位点上发生强吸附,从而抑制了环氧化反应的进行。因此,他们对之前提出的协同反应机理进行了修正,当环氧丙烷在Au-Ti 界面位点(S2)生成后,增加了环氧丙烷在金位点(S1)上的吸附这一基元反应步骤,如图6 所示。

图6 Au/TS-1催化剂上丙烯氢氧环氧化基元反应步骤Fig.6 The elementary reaction steps of HOPO reaction over Au/TS-1 catalyst
由该基元反应步骤推导出的环氧丙烷生成速率表达式如式(9)所示(L2为S2活性位点的总数目):

对考虑环氧丙烷强吸附的幂函数型动力学模型进行拟合,处理后得到氢气的反应级数为1±0.2,氧气的反应级数为0.4 ± 0.06,丙烯的反应级数为0.4±0.1,环氧丙烷的反应级数为-0.6± 0.2。通过对环氧丙烷生成速率和反应物分压的对数进行求导可建立反应物级数与覆盖度(A)之间的关系,如表2 所示,由此可推知HOOH 的覆盖度接近0,O2在S1吸附位点上的覆盖度约为0.6,C3H6在S2吸附位点上的覆盖度约为0.6,PO 在S1吸附位点和S2吸附位点上的覆盖度在0.2~0.4之间,且在两个吸附位点上的覆盖度之和为0.6。

表2 表面最丰物种的真实反应级数n与覆盖度A的关联[45]Table 2 Relation between the true reaction order and the coverages of the most abundant surface species[45]
根据以上分析可知基元反应步骤中所示的两个活性位点上各物种的覆盖度之和均满足小于1这一限定,这说明上述反应机理与实验得到的各反应物级数相契合,进一步验证了协同机理的合理性。此外,该课题组对生成水的幂函数型动力学模型进行拟合后得到氢气的反应级数为0.9 ±0.1,氧气的反应级数为0.3± 0.1,丙烯的反应级数为-0.3 ± 0.07,环氧丙烷的反应级数为0 ± 0.2,生成环氧丙烷和生成水的反应级数的差异预示着水和环氧丙烷并非在同一个活性位上生成,如图7所示。
1.3 包含H2O生成的反应动力学
HOPO 反应中,H2O 的生成对氢效有显著影响,进而影响过程的经济性。因此,阐明HOPO 反应中H2O 的生成路径对于提高催化性能至关重要。Oyama 等[43]首先在Au/Ti-TUD 催化剂上研究了H2O生成的动力学,提出了幂函数型的动力学方程,其中H2、O2、C3H6对水的生成级数分别为0.67、0.16、0.03,由此可知,氢气浓度对H2O 的生成速率影响最大,而丙烯对H2O 的生成几乎没有影响。这与Nijhuis 等[34]观察到的结果不同,其使用H2直接氧化作为探针反应,对比TiO2和SiO2载体负载Au纳米颗粒的反应情况,结果发现,共进料丙烯可以大大减小氢气的氧化速率,而氢气的直接氧化反应仅发生在Au上,由此可以认为丙烯的抑制作用归因于其在Au 位点与氢气发生了竞争吸附,由此提出丙烯在Au 位点的吸附显著减少了Au 上氢气氧化直接生成H2O 的数量,认为丙烯和氢气的竞争吸附减弱了Au位点上水的直接生成。他们首先使用微反应器研究了Au/Ti-SiO2上水和PO 的生成,将不同温度下PO 生成速率与水生成速率关联起来,如图8 所示,发现随着温度的升高,水生成的活化能并未出现明显改变,而PO 生成的活化能显著变化,进一步说明水的生成并不只出现在环氧化的产物中,而是通过两种路径生成:过氧物种的环氧化反应以及H2和O2直接反应。
他们进一步研究了在HOPO 反应体系中水在Au/Ti-SiO2催化剂上的生成路径及动力学[46],提出HOPO 反应中水有三种生成路径:(1)Au-Ti 位点上环氧化反应生成环氧丙烷时会生成水;(2)H2和O2在孤立的Au 位点以及Au-Ti 位点上均会直接生成H2O;(3)Au-Ti位点上用于丙烯环氧化的过氧化物中间体分解生成H2O,如图9 所示。基于这三种生成水的路径,在Au/Ti-SiO2催化剂上建立了PO 和水生成的动力学方程。
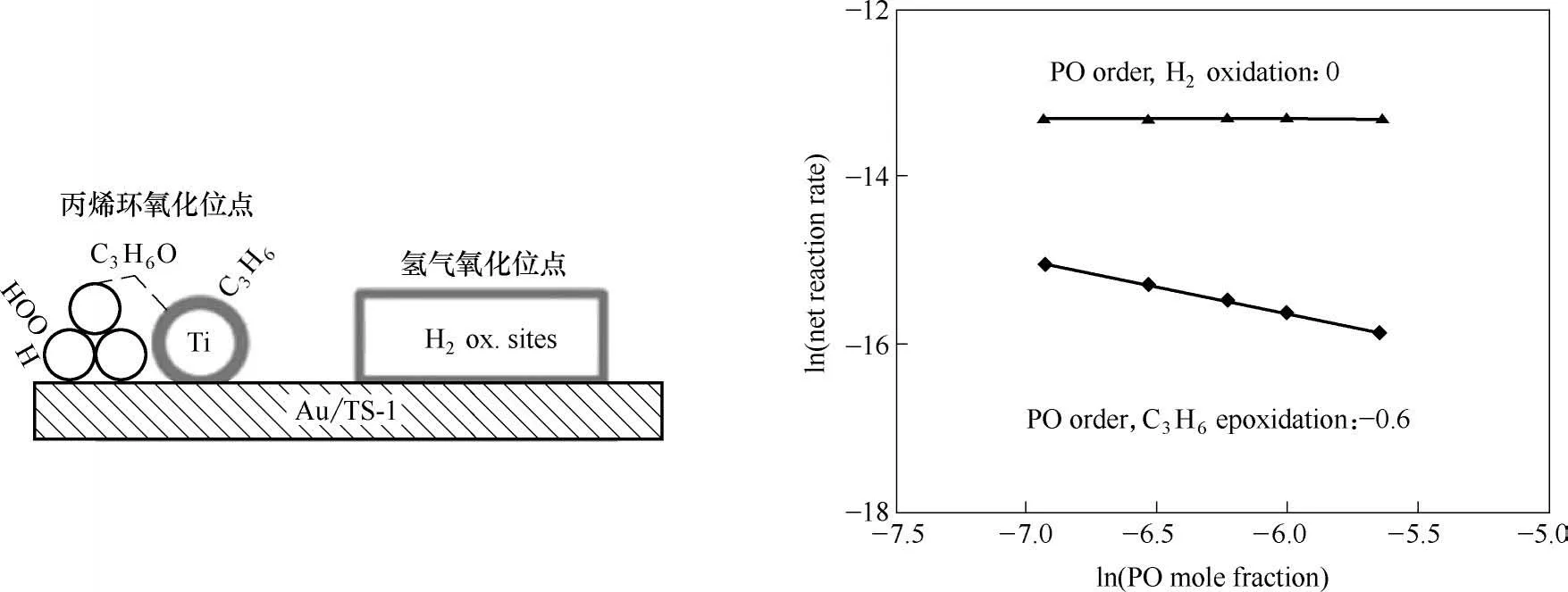
图7 Au/TS-1上丙烯氢氧环氧化反应中PO的负级数及共吸附示意图[45]Fig.7 The model for reaction of propene epoxidation with H2 and O2 over Au/TiO2[45]
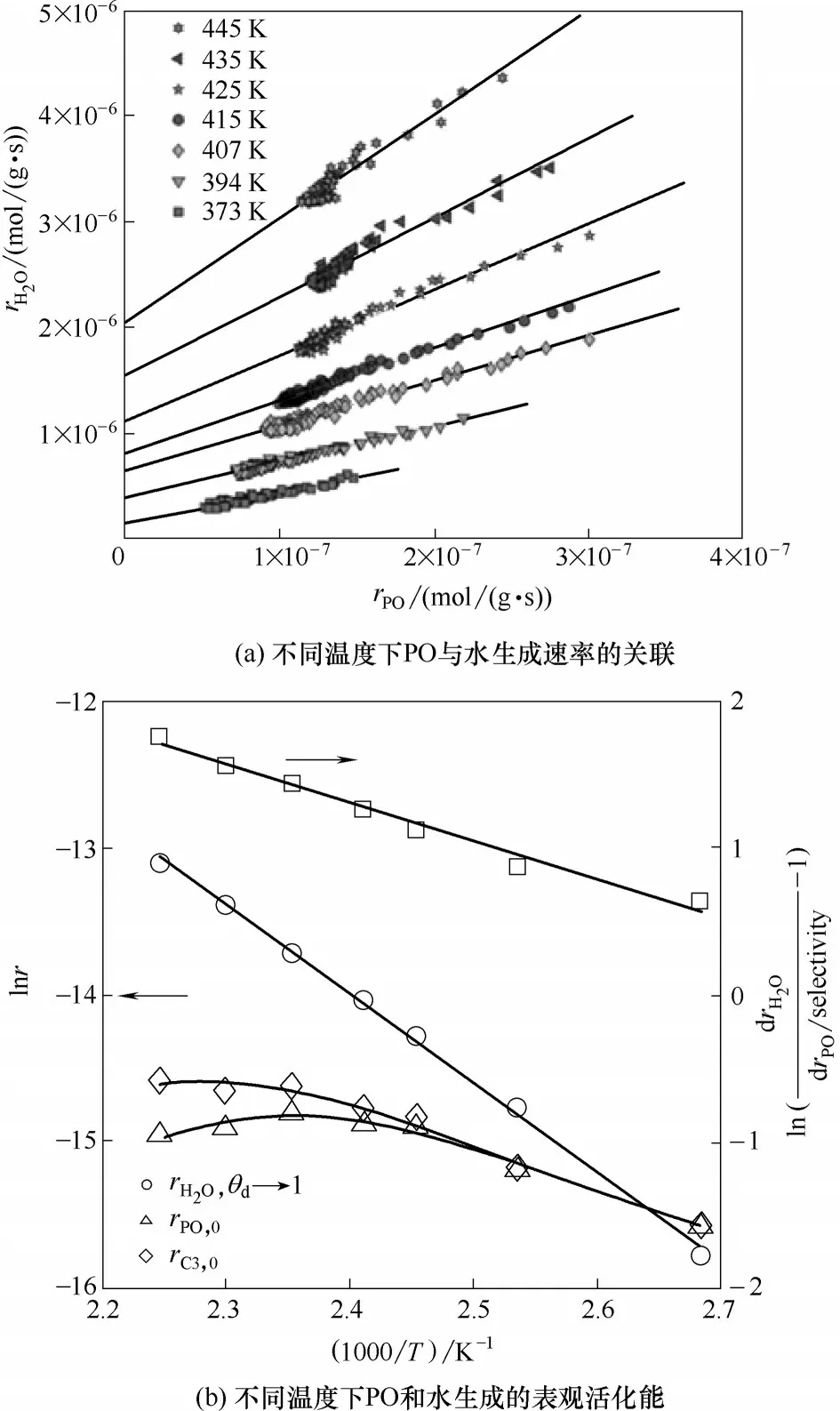
图8 Au/Ti-SiO2催化剂上水和PO生成的关联[37]Fig.8 The relation between water and PO formation in HOPO reaction over Au/Ti-SiO2 catalyst[37]
该机理假设本反应中有三个独立的位点,其中O2吸附在独立的Au 位点(S1),而氢气与丙烯在另一个Au 位点(S2)竞争吸附,HOOH 在Ti 位点(S3)与丙烯发生环氧化反应。在PO 生成的反应中,决速步骤为HOOH 物种的生成和环氧化反应,而在水生成的反应中,决速步骤分别为HOOH 的生成反应和HOOH物种的分解,如图10所示。

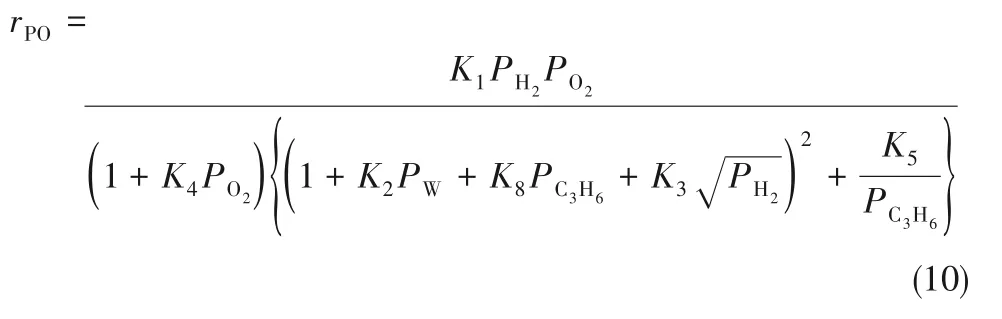


上述针对HOPO 反应动力学的研究为理解不同催化剂上HOPO 反应机理及后续反应器设计提供了重要的理论基础。然而,相关的动力学研究中使用了活性较低或稳定性较差的催化剂,且研究主要集中在生成PO、H2O 和CO2的反应,尚未涉及乙醛、丙酮、丙醛和丙烯醛等副产物的生成反应。因此,仍有待在高效稳定的催化剂上获得完整的反应网络及动力学,以用于指导该过程反应器的开发。
2 基于安全性与经济性的HOPO 反应器概念设计
HOPO 反应进料气体中既含有可燃性气体(即C3H6和H2),又含有助燃性气体(即O2),并且C3H6和H2在O2中有着很宽的爆炸极限范围(分别为2.0%~59%[47]和4.0%~95.2%[48]),因此进料气体在传统固定床反应器中的预混合及反应过程中存在爆炸风险[49],制约了其工业化应用。为了避免爆炸,通常会使用大量N2等惰性气体稀释进料气体,使混合气体远离爆炸极限范围。在已经报道的文献中,最典型的进料体积配比为C3H6/H2/O2/N2等于1/1/1/7。实验结果显示[9,24,50],该配比确实可以保证进料气体的安全性,但是较低浓度的反应物浓度限制了该反应的反应性能[51],如C3H6转化率<10%、PO 选择性<90%和PO 出口浓度<1.0%等[9]。同时,循环大量N2的过程必然会增加该过程的能耗,不利于过程的经济性[52]。针对该问题,有研究者提出反应器的概念设计应用于HOPO 反应,以同时满足该反应的安全性与经济性。
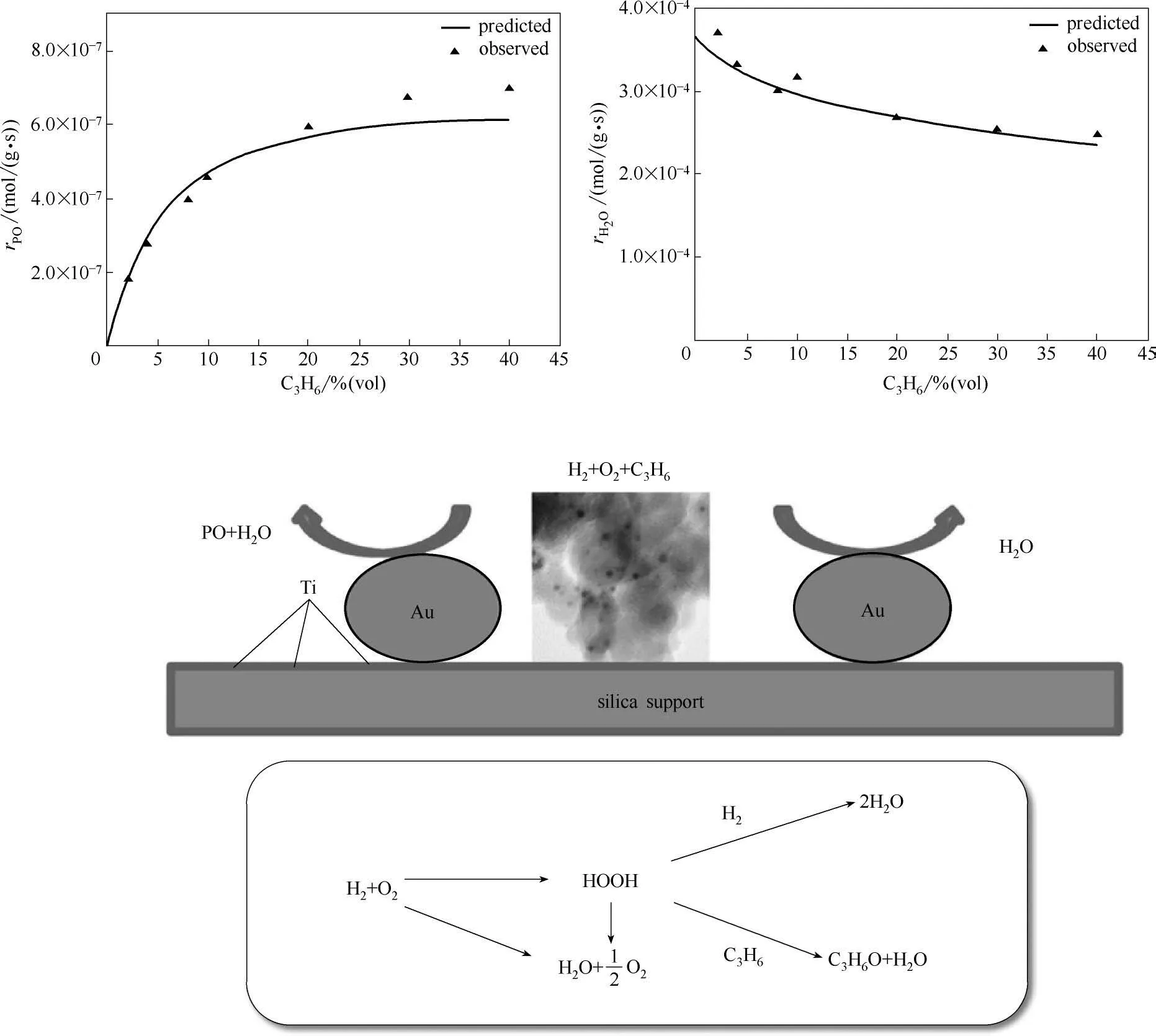
图9 Au/Ti-SiO2催化剂上HOPO反应中水的生成路径Fig.9 Routes for water formation in Au/Ti-SiO2 catalyzed HOPO reaction
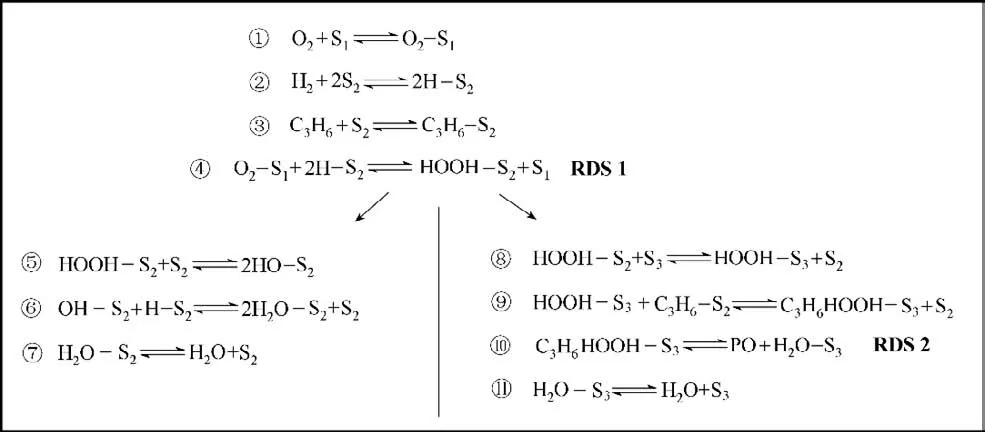
图10 Au/Ti-SiO2催化剂上包含水生成的HOPO基元反应步骤[46]Fig.10 The elementary reaction steps including water formation of HOPO reaction over Au/Ti-SiO2 catalyst[46]

图11 Au/Ti-SiO2催化剂上动力学结果预测值及实验值[46]Fig.11 Parity plot for experimental observations and kinetic expression for PO and water over Au/Ti-SiO2[46]
2.1 微通道反应器
微通道反应器有很小的体积,在这样的通道中发生爆炸产生的能量小于1 J,不会影响反应器的整体性。其次,微通道反应器有很好的控温系统,能够显著减少反应器中难以控制的风险。最重要的是,反应器的直径小于分子的平均自由程,说明反应通道内的火焰不太可能传播,分子会将它们的能量传播到管壁上而不是互相传递[53]。Nijhuis 等[36-37]使用0.9 mm 管径的微通道反应器,在排除了爆炸风险的基础上,使用Au/Ti-SiO2催化剂上研究了爆炸范围内的HOPO 反应动力学。Delgass 等[38]对Au/TS-1 催化剂的研究显示,PO 的生成速率主要取决于H2的浓度。但H2在O2中具有非常宽的爆炸极限,使用传统反应器时,必须严格控制H2的浓度。鉴于氢气在所有可燃性气体中的火焰直径最小[54](0.3 mm),Zhou等[55]提出使用电热微通道反应器,将通道尺寸控制在0.3 mm 以下,便可使用任何浓度的反应物,而不受爆炸影响[56]。该微通道反应器的装配如图12 所示,首先将TiO2镀于金属电热带的外层,再在上面涂刷Au/TiO2催化剂,之后将电热带折叠并置于套管中。使用该微通道反应器对丙烯环氧化反应进行测试,由于受到催化剂(Au/TiO2)本身活性的限制,该反应器能达到的反应性能非常有限,如丙烯转化率最高为1.7%,如图12所示。
2.2 H2选择性膜反应器
Oyama 等[57]报道的动力学为rPO=k[H2]0.54[O2]0.24×[C3H6]0.36,其中氢气的反应级数最高,他们认为提高氢气的量更能提高丙烯转化率和环氧丙烷的生成量。但是,由于氢气在纯氧气中的爆炸极限范围非常宽,在传统的反应器中提高氢气的含量会使进料气体浓度进入爆炸极限范围,产生爆炸的风险。为此,他们设计了氢膜反应器,该反应器中膜对氢气具有选择性[58]。如图13 所示,在该反应器中,氢气与丙烯、氧气和氩气分开进料,避免了氢气和氧气的直接接触,并且能够允许足够氢气从外侧的渗透进入反应区域,以提高氢气在反应过程中的浓度。结果显示,在较高的氢气浓度下,此反应器相比于传统的固定床反应器能够将环氧丙烷的生成速率提高1~2 倍。当反应温度从160℃升高到212℃时,C3H6转化率从6.5%增加到16%,而PO 的选择性从82%降低到65%。值得指出的是,此反应器能够显著提高丙烯的转化率,但是得到的PO 选择性远远低于工业要求目标。
2.3 催化膜反应器
对于HOPO 反应,除了C3H6转化率和PO 选择性,氢效(可表示为PO 与H2O 生成量的比值或者PO与H2转化率的比值)是评价该反应经济性的重要指标。实验表明,反应物的浓度对氢效有很大的影响:高的C3H6浓度有利于提高氢效,高的H2浓度虽然有利于PO 的生成,但会增加H2O 的生成,导致了氢效的降低,而针对膜反应器的操作优化有利于改善反应结果[59]。Nijhuis 等[60]采用催化膜反应器,研究了不同进料方式对氢效的影响程度,如图14 所示。Wicket-kallenbach 单元是研究流体在多孔介质中扩散的标准设备,该设备的特殊性在于多孔材料表面的两个孔的压力相等,流体的浓度梯度成为化合物扩散的驱动力。其中,3 为Au/Ti-SiO2催化膜,是丙烯、氢气和氧气的扩散和反应的区域。实验结果表明,第二种进料方式,即氢气与丙烯和氧气分开进料,能够有效地限制H2浓度,提高氢效。

图12 电热微通道反应器装配图及其反应性能[54]Fig.12 Microchannel reactor assembly and reactor performance[54]

图13 H2选择性膜反应器示意图其反应性能Fig.13 Schematic diagram of the H2-selective membrane reactor and reactor performance
另外,Nijhuis 等[60]建立了该膜催化反应器的模型,模拟结果显示:(1) 当膜厚度大于0.2 mm 时,能够确保膜反应器得到较高的氢效;(2)通过改变反应物通过催化膜阻力,使用足够的丙烯并控制氢气沿催化膜的扩散,有利于提高PO 的生成速率以及氢效。

图14 催化膜反应器示意图及进料方式Fig.14 Wicke-Kallenbach membrane reactor and different feeding strategies
值得指出的是,上述反应器的概念设计虽然保证了过程的安全性,但由于受到了催化剂性能或者反应物浓度的限制,并未表现出具有工业化前景的反应性能,即同时满足C3H6转化率>10%、PO 选择性>90%以及PO 出口浓度>1.0%。另外,相比于传统的固定床反应器,微通道反应器存在易堵塞、非均相催化剂不易配置等问题,而H2或者O2选择性膜反应器一般不适用于该反应,原因是该反应的反应温度为200℃左右,而H2或者O2选择性膜分别需要在300℃[61-65]和700℃[66-69]以上才能表现出可观的H2或者O2渗透通量。
2.4 H2/O2单独进料的膜反应器
Zhou 等[70]建立了四种不同进料方式反应器模型:固定床、二段床、H2和O2分开进料的膜反应器的二维拟均相模型,通过模拟计算比较了反应器内的温度和浓度分布,以及反应器出口C3H6转化率、PO选择性以及氢效。如图15 所示,研究结果表明,使用膜反应器可在反应器中保持较高的H2或O2浓度,有利于C3H6转化率和PO选择性,但也促进了H2O的生成,降低了氢效。O2单独进料的膜反应器避免了C3H6/H2和O2预混合的爆炸危险,更具有研究与开发价值。模拟研究表明,通过优化工艺操作条件,可同时达到C3H6转化率≥10%且PO 选择性≥90%的工业应用目标。

图15 O2/H2单独进料的膜反应器与传统反应器示意图及反应性能对比[70]Fig.15 Schematic diagram of the MR-O,MR-H,TSR,PBR and reactor performance[70]
3 总结与展望
近年来,针对Au-Ti 双功能催化剂催化HOPO体系的反应动力学及反应器概念设计的研究已经取得一定进展,主要包括多种类型Au-Ti 催化剂上由不同反应机理推导得到主反应的动力学模型,以及为实现反应过程安全高效进行而展开反应器概念设计,例如为避免爆炸风险而设计的微通道反应器、不同进料方式的膜反应器等,上述研究对于推进HOPO过程的工业化应用具有重要指导意义。但目前能满足工业要求的高稳定性与高活性兼顾的催化剂仍未出现,本体系距离工业化应用仍有差距。针对HOPO 反应的动力学与反应器设计研究,以下几方面工作值得进一步开展。
(1)目前,研究者主要通过多种表征手段和理论计算等来探究反应机理,但对副产物如丙醛、丙烯醛、丙酮、乙醛等生成途径的研究较少,且缺乏直观的实验现象证明上述产物的生成途径,有待结合表征手段、动力学辨析实验及微观动力学分析开展研究,以建立主副产物的反应网络。
(2)尽管当下针对不同的Au-Ti 催化剂性能开展了动力学及反应器研究,但针对目前最高性能的催化剂,仍缺少与之匹配的反应及失活动力学,并将其应用于传统固定床反应器中,以开展操作优化及流程设计。
(3)目前建立的反应动力学尽管能够很好地预测实验室条件下各自催化剂的反应结果,但均采用了粉末催化剂进行研究,但对工业成型催化剂,考虑到可能的内扩散影响和黏结剂等的作用,在进行工业反应器设计之前还需要利用动力学实验结果,耦合催化剂颗粒尺度与反应器尺度的传质传热过程,应用于工业反应器。
