光动力治疗中提高光敏剂靶向性的研究进展
2021-01-30杨宇鑫赵学泽樊江莉彭孝军
杨宇鑫,赵学泽,樊江莉,2,彭孝军
(1 大连理工大学精细化工国家重点实验室,辽宁大连116024; 2 大连理工大学宁波研究院,浙江宁波315016)
引 言
光动力治疗是一种基于光子诱发光敏剂产生活性氧的新型癌症治疗方法[1]。治疗过程如下:首先给患者局部或静脉注射光敏剂,经过一段时间后光敏剂在肿瘤组织中富集。然后使用特定波长的光照射肿瘤部位,光敏剂能够吸收光子产生大量的活性氧物种。活性氧物种攻击细胞,使生物分子和细胞发生形态或功能上的异变,造成直接的细胞损伤来致死细胞,从而杀死癌细胞并抑制其生长[2-4]。
传统的光敏剂对正常组织和肿瘤组织的区分能力有限,在杀伤肿瘤细胞的同时会对正常组织造成毒副作用[5]。当前光动力治疗研究的热点领域之一是如何进一步提高光敏剂的肿瘤靶向性[6-7]。构建靶标型光敏剂和可激活光敏剂是提高光敏剂肿瘤靶向性的有效策略。靶标型光敏剂是将光敏剂分子共价连接靶标基团,通过靶标基团特异性识别癌细胞,提高光敏剂的靶向能力。而可激活光敏剂能够被肿瘤微环境内特定的靶向因子激活,特异性识别并杀伤肿瘤组织。本文分别介绍了靶标型光敏剂和可激活光敏剂的研究进展,并对未来光动力治疗面临的挑战进行了展望。
1 光动力治疗的原理
光动力治疗(photodynamic therapy, PDT)是一种使用光敏剂(photosensitizer,PS)进行预处理,并在肿瘤区域使用激发光源照射,产生活性氧物种(reactive oxygen species, ROS)以破坏癌细胞的肿瘤治疗方法[8]。在特定波长的光源照射下,光敏剂会吸收光子,从基态(S0)跃迁到单重激发态(S1)[9]。激发态的分子极其不稳定,它可通过非辐射跃迁回到基态,放出热量;或通过辐射跃迁回到基态,发出荧光;也可以通过系间窜越(intersystem crossing,ISC)到达寿命相对较长的三重激发态(T1)。T1态光敏剂的两种失活途径对应了光动力治疗的两种机理(图1)。
一种是Ⅰ型反应机理:光敏剂在细胞微环境中和生物分子发生电子或质子转移,形成自由基。自由基与氧气发生电子转移,生成超氧阴离子自由基(O2-•),最终通过歧化作用或者单电子还原过程生成具有生物毒性的羟基自由基(OH•),导致细胞氧化性损伤[11-12]。另一种是Ⅱ型反应机理:三重激发态的光敏剂与基态氧(3O2)发生直接能量转移,生成毒性极强的单线态氧(1O2),从而导致癌细胞凋亡或坏死[13]。Ⅱ型机理的光敏剂在治疗过程中高度依赖氧气浓度,并且会消耗大量的氧气,加剧肿瘤乏氧程度。而研究表明,一些Ⅰ型PDT 即使在乏氧环境下也能表现出有效的治疗效果[14]。对于两种机理的光敏剂来说,活性氧量子产率都是决定光动力治疗效果的重要因素之一。
相比于手术切除、放疗、化疗等传统的癌症治疗方法,光动力治疗具有微创、极低的耐药性、低副作用和时空选择性高等优点。
2 光敏剂
光动力治疗的三大要素是激发光、氧气和光敏剂。其中,光敏剂是光动力治疗中最为关键的因素。
卟啉类光敏剂是光动力治疗中的第一代光敏剂。早在1975 年,血卟啉衍生物(HpD)就已成功用于光动力治疗。Photofrin[15]——从HpD 分离的卟啉二聚体和低聚物的混合物,已被批准用于浅层肿瘤的治疗。但是,该类光敏剂存在化学纯度低、组织穿透深度低、毒副作用大等缺点[16]。第二代光敏剂以酞菁类化合物、卟吩及其衍生物、竹红菌素等为代表[17-19]。第二代光敏剂具有更高的化学纯度,最大吸收波长红移至600~800 nm 范围内,可用于相对深层肿瘤的治疗[10]。
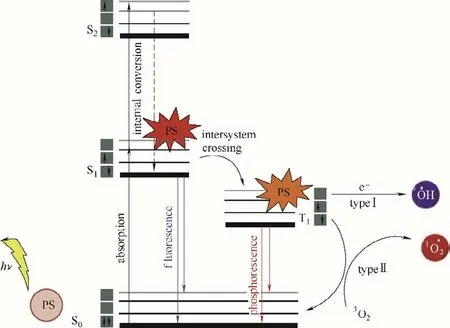
图1 光动力治疗的机理示意图[10]Fig.1 Schematic illustration of the mechanism of photodynamic therapy[10]
但是,传统的光敏剂对肿瘤的选择性有限,在光辐射条件下发挥光疗作用的同时,也会对周围的正常组织产生光毒性,严重限制了光动力治疗的临床应用。提高光敏剂的肿瘤靶向性以实现对肿瘤组织的特异性诊断与治疗,具有重要的意义。因此,目前正在发展的第三代光敏剂是在第二代光敏剂的基础上,将光敏剂与靶标基团共价连接,或者将光敏剂包载在纳米载体中,以提高光敏剂的肿瘤组织靶向性[20]。
理想的光敏剂应具有以下特征:(1)极低的暗毒性和高的光毒性;(2)在长波长处具有大摩尔消光系数;(3)对肿瘤组织具有高选择性;(4)优良的两亲性。表1介绍了目前常用的几种光敏剂母体。

表1 常用光敏剂母体简介Table 1 Introduction of commonly used fluorophores as photosensitizers
3 靶标型光敏剂
靶标型光敏剂是将光敏剂分子共价连接肿瘤靶标基团(如单克隆抗体[12]、多肽[21]、糖类等),利用靶标基团与癌细胞表面特异性受体结合靶向递送光敏剂,实现光敏剂对癌细胞的杀伤高于对正常细胞的损害[22]。当前研究常用的肿瘤靶标基团有抗癌药物、叶酸、生物素、核黄素等。下文从常用靶标基团进行分类,介绍了目前靶标型光敏剂的研究进展。
3.1 抗癌药物靶向
近年来,研究者们开发了许多将临床批准的化疗药物引入分子结构的光敏剂。将抗癌药物作为靶标基团与光敏剂共价连接,能够在提高光敏剂肿瘤靶向能力的同时,实现一定的协同化疗作用。
Xue 等[23]首次将锌酞菁与化疗药埃罗替尼共价连接合成了光敏剂3a、3b[图2(a)]。该类光敏剂以埃罗替尼作为靶向基团,实现了对表皮生长因子受体过表达癌细胞的选择性光动力治疗。其中,光敏剂3a 对HepG2(人源肝癌细胞)细胞的IC50(半抑制浓度)值为0.01 μmol/L。同时,埃罗替尼能够改善光敏剂的两亲性和生物相容性。随后,基于此思想,研究者们对抗癌药物修饰的靶标型光敏剂进行了广泛的研究。
Jung 等[24]将抗血管生成抑制剂乙酰唑胺(AZ)作为靶标基团,与传统的BODIPY 类光敏剂BPS 共价连接,合成了光敏剂AZ-BPS[图2(b)]。AZ-BPS的乙酰唑胺部分能够靶向癌细胞中过表达的碳酸酐酶IX(CAIX),并对CAIX 产生抑制作用,进而抑制肿瘤血管的生成[25-26],提高PDT 对乏氧肿瘤的治疗效率;而BPS 部分在660 nm 波长的光激发下,能够发出荧光并产生单线态氧,杀伤癌细胞。活体实验表明,与单纯的BPS 相比,AZ-BPS 对碳酸酐酶阳性肿瘤的选择性得到了大幅提高,与各种血管生成因子相关的基因表达得以下调。但是,体外和体内实验中使用的光剂量(3600 J/cm2)远高于临床使用标准。
在肿瘤细胞中,热休克蛋白90(Hsp90)过量表达。药物Ganetespib 能够靶向Hsp90 并抑制其表达,从而有效抑制肿瘤细胞的增殖并诱导细胞凋亡[27-29],具 有 优 秀 的 肿 瘤 选 择 性。Huang 等[30]将Ganetespib 与锌酞菁光敏剂ZnPc 共价连接,制备了光敏剂Gan-ZnPc[图2(c)]。光敏剂Gan-ZnPc 能够选择性识别并结合癌细胞表面的Hsp90,从而在癌细胞中富集,并通过Hsp90 介导的细胞内化作用进入癌细胞,产生大量的活性氧杀伤肿瘤组织。在MCF-7(人源乳腺癌细胞)细胞体外实验和带有4T1(鼠源乳腺癌细胞)细胞异种移植物的小鼠活体实验中,Gan-ZnPc均显示出强大的肿瘤选择性和抗肿瘤作用。
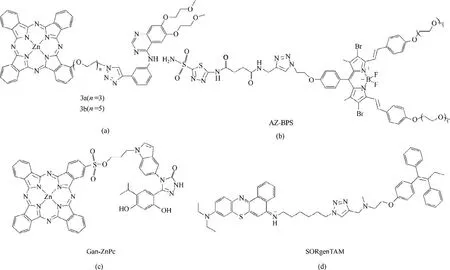
图2 3a、3b、AZ-BPS、Gan-ZnPc和SORgenTAM的化学结构示意图Fig.2 Schematic illustration of the chemical structure of 3a,3b,AZ-BPS,Gan-ZnPc and SORgenTAM
目前报道的光动力治疗系统大多数是通过高度依赖氧气的Ⅱ型机理进行的[31]。但是,由于癌细胞的过度增殖和肿瘤中血液供应不足,氧气浓度随实体瘤中的位置不同而变化,某些区域氧含量非常低[32]。此外,Ⅱ型光动力治疗过程中会消耗氧气,同时阻塞肿瘤血管,造成更严重的乏氧,不利于肿瘤的治疗[33]。
抗雌激素药物他莫昔芬(TAM)能够通过影响线粒体电子传输来干扰细胞能量代谢过程,降低细胞内源性O2消耗速率并下调HIF-1α 蛋白的表达,从而减轻肿瘤的固有低氧环境[34]。本课题组[35]将TAM 引入光敏剂分子结构中,合成了一例基于Ⅰ型反应机理的光敏剂SORgenTAM[图2(d)]。实验结果表明,即使在严重的乏氧环境下,SORgenTAM 也能降低内源性O2消耗,并且利用节约的内源性O2生成O2-•,启动低氧气依赖性的Ⅰ型PDT。在低氧环境下(2% O2),较低浓度的SORgenTAM(0.63 μmol/L)在低剂量光照下(660 nm,20 mW/cm2, 10 min)就能够杀死约83%的癌细胞。
3.2 靶向癌细胞表面过量表达物
目前,利用癌症表面特异性受体与化合物分子给体之间的特异性结合是实现光敏剂靶向递送的主要方式[22]。
生物素受体在大多数肿瘤细胞表面是过表达的[36]。本课题组[37]将生物素作为靶标基团引入光敏剂分子中,合成了能够靶向生物素受体阳性肿瘤的光敏剂ENBS-B[图3(a)]。实验表明,即使在乏氧环境下(2%O2),ENBS-B 也能通过Ⅰ型反应机理产生大量的O2-•,并通过超氧化物歧化酶(SOD)介导的级联反应将部分O2-•转化为高生物毒性的OH•。这些自由基能够协同破坏细胞内溶酶体,从而触发癌细胞凋亡,表现出强大的低氧PDT 效果。受益于生物素配体的靶向能力,ENBS-B 在癌细胞中的细胞摄取率比在正常细胞中高87倍。
随后,本课题组[38]开发了另一例将生物素作为靶向配体的近红外光敏剂Se-Biotin[图3(b)]。Se-Biotin 能够有效区分肿瘤细胞(MCF-7 细胞)和正常细胞,从而显著降低对正常细胞的副作用。在660 nm波长的光源照射下,Se-Biotin在生物素受体阳性肿瘤中的IC50值低至85 nmol/L,表现出有效的抗癌效果。
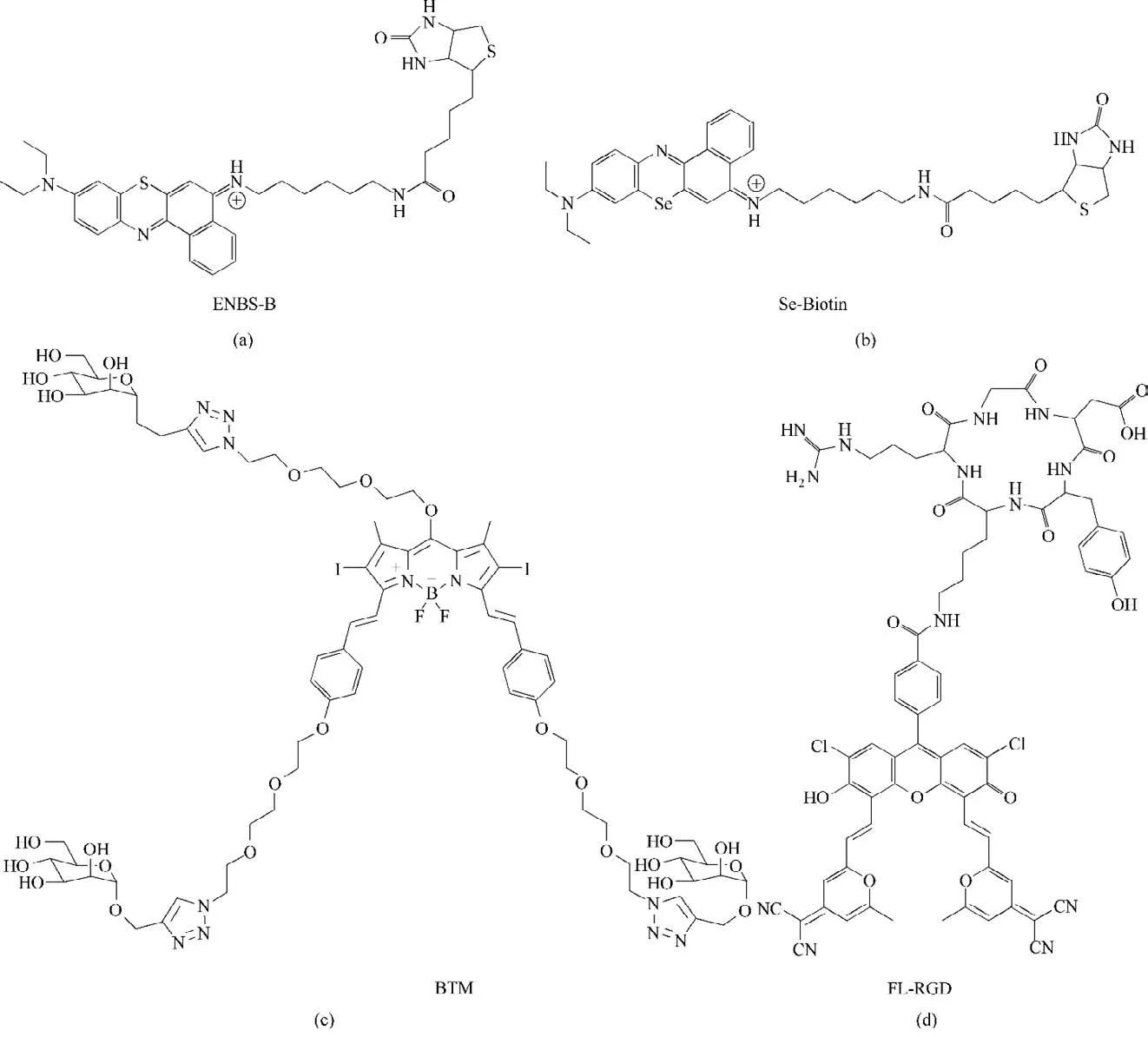
图3 ENBS-B、Se-Biotin、BTM和FL-RGD的化学结构示意图Fig.3 Schematic illustration of the chemical structure of ENBS-B,Se-Biotin,BTM and FL-RGD
甘露糖受体在多种癌细胞表面过表达。甘露糖修饰的光敏剂可通过甘露糖受体介导的内吞作用内化到癌细胞中。基于此思想,Zhang等[39]合成了与甘露糖共价连接的BODIPY 类光敏剂BTM,然后将BTM 与Tween 80 共组装为纳米胶束BTM-NMs[图3(c)]。BTM-NMs 能够通过高通透性和滞留效应(enhanced permeablity and retention effect,EPR)促进其在肿瘤部位的积累[40]。此外,将甘露糖连接在纳米载体表面能够增强BTM-NMs 的肿瘤靶向能力,减少其对正常细胞的副作用。BTM-NMs 通过甘露糖受体介导的内吞作用选择性内化到MDA-MB-231(人源乳腺癌细胞)细胞后,在细胞溶酶体中分解为光敏剂BTM。使用12 μg/ml 的BTM-NMs 和光源(665 nm,20 mW/cm2,30 min)照射进行光动力治疗后,癌细胞死亡率达到90%。
整联蛋白αvβ3是一种异质二聚体细胞表面受体,在多种癌细胞中过表达,但在大多数正常组织和血管中很少表达,它对肿瘤的生长、转移和血管生成起重要作用。精氨酸-甘氨酸-天冬氨酸三肽(RGD)是整联蛋白的识别序列,对整联蛋白具有选择性。Ranyuk 等[41]将锌酞菁与RGD 偶联,制备了ZnPc-RGD 偶联物。该光敏剂表现出与整联蛋白结合的特性,但光动力治疗效果较差。
研究表明,多聚的RGD 更有利于其与整联蛋白结合。因此,Liu 等[42]使用cycloRGD 修饰荧光素衍生物,制备了光敏剂FL-RGD[图3(d)]。FL-RGD 可以靶向肿瘤组织并进一步定位肿瘤细胞的溶酶体。在光源(630 nm,40 mW/cm2,20 min)照射下,FLRGD 处理的整联蛋白αvβ3阳性肿瘤的细胞凋亡率为46.6%。
3.3 结构固有靶向
癌细胞膜表面通常呈较高负电性。当光敏剂分子结构中带有正电荷时,光敏剂能够通过静电吸引作用靶向并进入癌细胞,增强光敏剂的肿瘤靶向能力[43-44]。
本课题组[45]报道了一种将罗丹明染料(RDM,λabs,max= 550 nm)作为能量供体,BODIPY 光敏剂(BDP,λabs,max=660 nm)作为能量受体的基于荧光共振能量转移(fluorescence resonance energy transfer,FRET)的光敏剂RDM-BDP[图4(a)]。由于分子结构中带有正电荷,RDM-BDP 结构固有靶向呈较高负电性的癌细胞膜,并且定位于线粒体。因此,RDMBDP 的肿瘤细胞摄取率比单纯的BDP 高了13 倍。并且,RDM 的发射波长与BDP的吸收波长具有很大的重叠。因此,当RDM 受到光激发后,其激发态能量能够有效传递给BDP。相对于BDP 的窄光谱范围而言,RDM-BDP 的吸收光谱得到明显拓宽(500~700 nm),从而提高了RDM-BDP 的1O2产生效率。在660 nm 激发光照射下,RDM-BDP 的1O2产生效率与BDP 相当。但是,在550 nm 激发光照射下,RDM-BDP 的IC50值仅为BDP 的1/81。经400~700 nm 宽谱光源照射后,0.16 μmol/L 的RDM-BDP 对肿瘤细胞的杀伤率大于90%,而BDP 仅为20%,RDMBDP的光动力效果与BDP相比得到明显提升。
随后,本课题组[43]报道了一例用于减轻光动力治疗氧气依赖的I 型光敏剂ENBOS[图4(b)]。通过FRET 过程,无光敏化性质的能量供体(荧光团ENBO)将其自身光谱吸收性质附加给能量受体(光敏剂ENBS),导致ENBOS在近红外光区的吸收强度明显加强,其O2-•量子产率得到提高。受益于ENBOS 净带两个正电荷,它具有强大的癌细胞靶向能力。在静脉注射48 h 后,ENBOS 的信噪比达到25.2。由于ENBOS 可通过Ⅰ型机理高效产生O2-•,ENBOS 能够有效杀伤乏氧肿瘤细胞。在乏氧实体瘤治疗实验中,ENBOS 的肿瘤生长抑制(TGI)率高达84%。

图4 RDM-BDP、ENBOS、ENBS和ENBO的化学结构Fig.4 The chemical structure of RDM-BDP,ENBOS,ENBS and ENBO
4 可激活型光敏剂
肿瘤组织具有不同于正常组织的微环境、酶和核酸等,例如:(1)肿瘤组织的细胞外pH(在6.0~6.8)通常低于正常组织(pH 为7.4 左右),呈微酸性[46-47];(2)肿瘤组织中谷胱甘肽(GSH)浓度通常高于正常组织[48];(3)肿瘤组织中某些酶活性异常,如过表达的碱性磷酸酶、硝基还原酶、偶氮还原酶和γ-谷氨酰转肽酶等[49]。这些特异性是可激活型光敏剂识别癌细胞的刺激源。
可激活型光敏剂(activatable photosensitizers,aPSs)具备刺激响应性质,它能够被特定的靶向因子激活以调控单线态氧的产生,选择性识别并杀伤肿瘤组织。在正常组织中,即使在光照射下,可激活光敏剂也保持猝灭的光失活状态,不会对正常细胞产生毒副作用。但是当aPSs 转运到靶组织后,靶向因子(如肿瘤微环境、酶、核酸等)将其选择性激活,从而选择性杀死肿瘤细胞[50]。开发近红外光激发的、能够彻底猝灭其基底活性氧的可激活型光敏剂是目前aPSs 的研发目标。下文从可激活光敏剂的猝灭机理进行分类,介绍当前的研究进展。
4.1 基于光诱导电子转移猝灭
光诱导电子转移(photoinduced electron transfer,PET)过程能够猝灭aPSs的激发态,从而猝灭其基底活性氧[51]。在肿瘤组织中,aPSs 的分子结构发生变化(例如与H+结合发生质子化反应,靶标基团被靶标分子切断等),导致PET 过程被抑制,从而恢复aPSs的单线态氧产生和荧光发射。
Chen等[52]报道了一种将锌酞菁ZnPc与2,4,6-三(N,N-二甲基氨基甲基)苯氧基(TAP)共价连接的pH 响应型可激活光敏剂ZnPc(TAP)4[图5(a)]。氨基通过PET 过程猝灭ZnPc(TAP)4在正常组织中的光活性。在生理pH 下,ZnPc(TAP)4无明显的光毒性。当缓冲介质的pH 从7.4 降低至6.5 时,氨基发生质子化反应,PET 过程被抑制,ZnPc(TAP)4的荧光强度和单线态氧产生效率均得到增强。在带有4T1细胞的小鼠活体治疗实验中,ZnPc(TAP)4不仅消融了肿瘤细胞,还能对肿瘤部位进行荧光成像,实现诊断与治疗一体化。

图5 基于PET的可激活光敏剂猝灭机理示意图Fig.5 Schematic illustration of quenching mechanism of PET-based activatable photosensitizer
Sun 等[53]利用带有正电荷的三苯基膦作为线粒体靶向配体[54-55],合成了谷胱甘肽与H2O2双重响应的可激活光敏剂mitoaPs[图5(b)]。在正常组织中,mitoaPs 的光毒性被彻底猝灭。在肿瘤组织中,mitoaPs与H2O2和谷胱甘肽发生反应,转化为具有光毒性的光敏剂。激活的光敏剂可通过Ⅰ型和Ⅱ型两种反应机理生成O2-•和1O2,减轻PDT 的氧气依赖程度。无 论 在21% O2还 是1% O2条 件 下,使 用H2O2(50 μmol/L)和mitoaPs(10 μmol/L)处理Hela(宫颈癌细胞)细胞24 h 后,再用激发光(405 nm, 30 mW/cm2)照射5~6 min,癌细胞存活率都低于10%。
Radunz 等[56]报道了一种基于酚羟基调控的pH响应型BODIPY类可激活光敏剂[图5(c)]。在正常生理pH下,去质子的酚羟基通过PET过程猝灭光敏剂的单线态氧和荧光的产生。在肿瘤组织的弱酸性条件下(pH =5.5),酚羟基发生质子化反应,PET 过程被抑制,导致光敏剂的荧光恢复,产生单线态氧。将HeLa 细胞与0.1 μmol/L 的光敏剂共孵育24 h 后,使用532 nm 的激光二极管照射5 min,肿瘤细胞几乎完全被杀死。但该BODIPY 类光敏剂的水溶性有待改善。
光敏剂的细胞摄取水平对光动力抗肿瘤效果有很大的影响[57]。Akkaya等[58]报道了一种GSH 响应的BODIPY 类可激活光敏剂PS1[图5(d)]。由于聚乙二醇链的引入,PS1 的细胞渗透性和水溶性得到了提高,从而提高了其细胞摄取水平。在正常组织中,2,4-二硝基苯磺酸基团通过PET 过程猝灭PS1的单线态氧和荧光的产生。在肿瘤组织中,过表达的GSH 将2,4-二硝基苯磺酸基团切断,PS1 的光敏性得到恢复。细胞毒性实验显示,在625 nm 的红光LED 照射下,PS1 对正常MRC-5(人胎肺成纤维细胞)细胞系没有明显的毒性,但对HCT116(结肠癌细胞)细胞系具有显著的光毒性,IC50值仅为20 nmol/L。得益于其较高的细胞摄取水平,PS1 仅在低浓度下即可高效杀伤肿瘤细胞。
目前报道的pH 激活的光敏剂大多需要在pH ≤5.5 时才能发挥作用。然而,肿瘤组织细胞外pH 通常在6.0~6.8。因此,制备能够在微酸性环境下被显著激活的光敏剂具有重要意义。
4.2 基于抑制分子内电荷转移猝灭
基于抑制分子内电荷转移(intramolecular charge transfer, ICT)的可激活光敏剂的调控机理如下:在正常组织中,aPSs 的ICT 过程被抑制。此时,激发态能量主要通过振动弛豫释放,aPSs 的系间窜越过程被阻止,从而限制aPSs 在正常组织中的单线态氧产生能力,使其光毒性最小化[59-62]。而在肿瘤组织中,aPSs被特异性激活转化为D-π-A结构的光敏剂,ICT 过程恢复,从而恢复光敏剂的单线态氧产生和荧光发射。
Liu 等[63]通过双硼酸酯键将抗癌药5′-DFUR 和半菁光敏剂(NPS)共价连接,开发了一种H2O2响应的近红外前药PNPS,用于近红外荧光成像和化疗协同光动力治疗[图6(a)]。在正常组织中,原光敏剂母体的羟基被苯硼酸基团取代,ICT 过程被抑制,从而降低PNPS 的光毒性。在肿瘤组织中,过表达的H2O2可破坏双硼酸酯基团,释放出光敏剂NPS 和化疗药5′-DFUR。在白光照射下,PNPS 对Hela 细胞的IC50值为9.32 μmol/L。然而,Hela 细胞存活率小于10%时所需要的PNPS 浓度高达30 μmol/L,治疗效果较差,对正常组织的损伤很大。这可能是由于NPS中溴原子的取代位置不当,重原子效应弱,导致NPS的单线态氧量子产率较低。
本课题组[64]利用苄硝基干扰半菁染料的ICT 过程,合成了一种缺氧激活的近红外可激活光敏剂ICy-N[图6(b)]。ICy-N 的母体结构与PNPS 相同。不同的是,通过将碘原子引入吲哚环,改善了ICy-N的系间窜越过程,从而提高了ICy-N 的单线态氧量子产率。由于引入了硝基还原酶识别基团(苄硝基),ICy-N 的ICT过程被抑制,大大降低了ICy-N 对正常组织的光毒性。传统的菁染料斯托克斯位移较小[65]。但ICy-N 的最大吸收波长位于660 nm 处,最大发射波长位于710 nm 处,具有较大的斯托克斯位移。在肿瘤组织中,过表达的硝基还原酶将ICy-N 转化为ICy-OH,ICT 过程恢复。在乏氧条件下(10% O2),经光源(660 nm,12 mW/cm2)照射后,ICy-OH 可高效产生单线态氧杀伤肿瘤,IC50值仅为0.63 μmol/L。

图6 PNPS、ICy-N、APN-CyI和ALPPS的猝灭机理示意图Fig.6 Schematic illustration of the quenching method of PNPS,ICy-N,APN-CyI and ALPPS
随后,本课题组[66]将氨基肽酶N(APN)响应基团引入半菁染料CyI-OH中,制备了APN激活的近红外可激活光敏剂APN-CyI[图6(c)]。使用2 μmol/L 的APN-CyI 进行治疗,在光源(660 nm,20 mW/cm2,20 min)照射下,癌细胞存活率仅为15%,而正常细胞存活率为70%,进一步降低了可激活光敏剂对正常组织的光毒性。
Zhai 等[67]开发了一种高效的长波长D-π-A 光敏剂PSSe-I,并使用磷酸酯基团将光敏剂PSSe-I磷酸化,制备了彻底猝灭型可激活光敏剂ALPPS[图6(d)]。PSSe-I的pKa值为5.78,适用于生理pH 环境。ALPPS 的ICT 过程被抑制,导致ALPPS 在可见光处的吸收彻底消失,光活性被彻底抑制。在肿瘤组织中,过表达的碱性磷酸酶将磷酸酯基团切断,ALPPS 转化为PSSe-I,ICT 过程恢复,光活性得以恢复。PSSe-I的最大吸收波长位于616 nm 处。使用5 μmol/LALPPS 处理Hela 细胞并在氙气灯(490~700 nm,10 mW/cm2,10 min)的照射下,Hela 细胞存活率小于13%。由于ALPPS的光活性被彻底猝灭,ALPPS对正常细胞的损伤极低,表现出彻底猝灭型可激活光敏剂的优势。
4.3 基于罗丹明衍生物分子内螺环化猝灭
大部分的可激活光敏剂设计策略是通过光诱导电子转移过程或者抑制分子内电荷转移效应。然而,通过这两种策略难以彻底猝灭可激活光敏剂的基底活性氧,而少量的活性氧足以对正常组织造成破坏。因此,如何彻底抑制可激活光敏剂在正常组织中的光毒性是研究的重点。罗丹明衍生物处于闭环状态时无光活性,能够实现可激活光敏剂基底活性氧的彻底猝灭[68]。当螺环一旦被打开,罗丹明即可恢复光活性。
Lv 等[69]报道了一种罗丹明类可激活光敏剂Se-NR-Az[图7(a)]。Se-NR-Az 没有光毒性,其基底活性氧能够被彻底猝灭。当Se-NR-Az 与外源性三苯基膦(TPP)发生Staudinger 反应后[70-72],Se-NR-Az 会转化为具有光毒性的光敏剂Se-NR。将Hela 细胞与Se-NR-Az 和200 μmol/L 三苯基膦共孵育,并使用光源(610 nm,6 J/cm2)照射,Se-NR-Az 的IC50值仅为0.205 μmol/L。但是,Se-NR-Az 进行光动力治疗时需要外加非细胞内源性的三苯基膦,无法控制Se-NR-Az 在正常细胞中不与三苯基膦发生反应转化为Se-NR,肿瘤选择性较差。

图7 罗丹明类可激活光敏剂激活机理示意图Fig.7 Schematic illustration of the activation method of rhodamine-based activatable photosensitizers
Urano 课题组[73]将β-半乳糖苷引入罗丹明母体,合成了一例β-半乳糖苷酶激活的光敏剂HMDESeR-βGal。HMDESeR-βGal 无光活性,单线态氧产生被彻底猝灭。在β-半乳糖苷酶过表达的细胞中,HMDESeR-βGal与β-半乳糖苷酶反应转化为HMDESeR,恢复其在可见光处的吸收和单线态氧产生能力,从而诱导细胞死亡。
随后,Urano 课题组[74]报道了另一例γ-谷氨酰转肽酶(GGT)响应的罗丹明类可激活光敏剂gGlu-HMSeR[图7(b)]。在正常组织中,gGlu-HMSeR 无光活性,其在可见光处的吸收被彻底猝灭,从而彻底猝灭了其单线态氧的产生。在带有肿瘤的鸡绒膜尿囊膜模型中,gGlu-HMSeR 会被过表达的GGT 特异性裂解开环转化为具有光毒性的光敏剂HMSeR,选择性杀伤肿瘤而不损害周围的健康组织。gGlu-HMSeR 的最大吸收波长位于532 nm 处。使用10 μmol/L 的gGlu-HMSeR 与SHIN3 细胞(人卵巢癌细胞)共孵育,经过光源(510~550 nm,50 mW/cm2,1 min)照射后,SHIN3细胞存活率小于20%。
综上所述,虽然基于罗丹明类螺环化的可激活光敏剂,在闭环状态下能够彻底猝灭其基底活性氧,但是,以罗丹明为母体的光敏剂最大吸收波长较短,通常<600 nm,组织穿透深度低,无法对深层肿瘤进行杀伤。因此,开发近红外光激发的、彻底猝灭型可激活光敏剂至关重要且面临巨大挑战。
5 结 论
构建靶标型光敏剂和可激活型光敏剂能够有效地提高光敏剂的肿瘤靶向性,极大地降低光动力治疗的毒副作用。但在临床治疗中,光动力治疗仍面临许多挑战。以下几个方面的问题在未来应予以解决。
(1)目前大多数的光动力治疗均基于Ⅱ型机理,对肿瘤内氧气浓度具有很大的依赖。因此,开发基于I型机理的可激活光敏剂具有重要意义。
(2)光敏剂是否具有极低的暗毒性,能否在体内循环中尽可能少地富集在正常组织和主要器官中,是其能否用于临床治疗的关键因素。在荧光染料中引入重原子是设计光敏剂的主要方法之一,但重原子会使分子的暗毒性增加。因此,开发无重原子、长三线态寿命的光敏染料是研究的重点。
(3)近年来,光热治疗、免疫治疗等治疗方法迅速发展。将光动力治疗与光热治疗、免疫治疗进行协同治疗,能够发挥多重治疗效果,有利于推动光动力治疗的发展。
