综合线性定量指纹法评价退热解毒注射液融合指纹图谱研究
2021-01-29兰丽丽巩丹丹孙国祥
章 越,兰丽丽,巩丹丹,孙国祥
(沈阳药科大学 药学院,辽宁 沈阳 110016)
退热解毒注射液是一种提取自中药的现代制剂,由金银花、连翘、牡丹皮、蒲公英、金钱草、柴胡、夏枯草、石膏8味药材组成,用于病毒性感染、不明原因高热、急慢性炎症等,特别适用于抗生素耐药或过敏患者[1]。由于其疗效确切、作用快速、生物利用度高,故广泛应用于临床。据报道,在此次新型冠状肺炎疫情中,退热解毒注射液也发挥了重要的治疗作用[2-3]。目前,中药部颁标准中仅采用绿原酸含量作为其质量控制指标,已有文献报道[4-6]也多基于1种或几种有效成分的定量分析进行质量控制,但对于成分多样、机制复杂的中药制剂来说,这种控制模式显然是不够完善的。近年来,指纹图谱技术由于其整体性、模糊性、可量化等特点,已成为国内外广泛接受并收载于各国药典及相关法规中的中药质控最有效的综合模式之一[7-9]。但指纹图谱建立后,如何利用其丰富的化学信息来全面评价样品质量以及获取相关的药效活性信息极为关键。目前,色谱指纹图谱的相似度评价方法有相关系数法、夹角余弦法、Nei系数法等[10-12],但均以定性评价为主,难以展示样品的整体化学成分含量差异。本文通过建立多波长融合指纹图谱来全面表征样品的化学成分信息,并提出了一种新的相似度评价方法——综合线性指纹法,从整体模式上定性定量评价不同批次样品的质量。该方法提供了综合线性定性相似度(Sl)、综合线性定量相似度(Pl)和指纹变异系数(α)3个主要参数,可为有效控制药物质量提供有益参考。
1 实验部分
1.1 综合线性指纹法的理论

(1)
(2)
(3)
(4)
(5)
(6)

(7)
1.2 仪器与试剂
安捷伦1100型液相色谱仪(配在线脱气机、四元梯度泵、二极管阵列检测器、自动进样装置),Agilent Chemstation(Edition C.01.07)网络工作站;乙腈和甲醇(色谱纯,山东禹王和天下新材料有限公司);磷酸(色谱纯,天津科密化学试剂公司);庚烷磺酸钠(山东禹城市中美色谱产品厂);1,1-二苯基-2-苦肼基自由基(DPPH,奥科鼎盛生物技术有限公司);实验所用去离子水及其他化学品均为分析纯级。退热解毒注射液(S1~S10)、退热解毒中间体(S11~S20)由西安高科陕西金方药业有限公司提供;绿原酸(CHA,批号:110753-201817)、连翘酯苷A(FA,批号:111810-201606)、丹皮酚(PA,批号:110708-201407)、柴胡皂苷D(SD,批号:110778-201711)标准品均购自中国食品药品检定研究院。
1.3 溶液配制
样品溶液:取退热解毒注射液适量,摇匀,过0.45 μm滤膜,取续滤液即得。对照品溶液:分别称取绿原酸、连翘酯苷A、丹皮酚、柴胡皂苷D的对照品适量,精密称定,加无水甲醇溶解,超声并定容至刻度,得质量浓度分别为160、200、60、200 μg/mL的对照品溶液。
1.4 色谱条件
COSMOSIL C18柱(250 mm×4.6 mm,5.0 μm)色谱柱;流动相:A为0.2%磷酸-水(含5 mmol/L庚烷磺酸钠)溶液,B为乙腈-甲醇(9∶1);梯度洗脱程序:0~10 min,96%~89%A;10~35 min,89%~80%A;35~45 min,80%~55%A;45~55 min,55%~45%A;55~70 min,45%A。流速1.0 mL/min,柱温为(35±0.15) ℃。检测波长为210、254、265、330、360 nm;进样量5 μL。
2 结果与讨论
2.1 指纹图谱分析
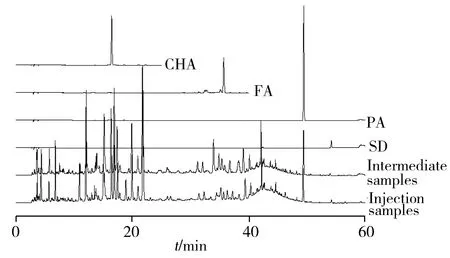
图1 退热解毒注射液和中间体样品代表指纹图谱 及对照品色谱图Fig.1 Representative fingerprints and chromatograms of the reference substance of Tuire Jiedu injection and intermediate samples
2.1.1 方法学验证取S1供试品溶液、各对照品溶液分别进样5 μL,记录色谱图,通过对比保留时间和在线紫外光谱定性,考察系统适用性(图1),由于图中绿原酸峰强度适中且与相邻峰分离较好,因此选其为参照物峰。精密吸取S1供试品溶液5 μL,连续进样6针,记录色谱图,考察方法的精密度。结果显示,以绿原酸的保留时间和峰面积为参照,各指纹峰的相对保留时间的相对标准偏差(RSD)均小于1%,相对峰面积的RSD均小于3%,表明系统进样精密度良好;平行制备6份S1供试品溶液,每份平行测定2次,记录色谱图考察方法的重复性。结果共有峰相对保留时间的RSD均小于2%,相对峰面积的RSD小于3%,表明方法的重复性良好;对同一S1供试品溶液,分别在0、5、10、15、25 h测定5次,记录色谱图,考察方法的稳定性。结果各指纹峰相对保留时间的RSD均小于1%,相对峰面积的RSD均小于3%,表明溶液的稳定性良好。由此可见,本研究所建立的分析方法适用于指纹图谱分析。
2.1.2 五波长融合指纹图谱建立众所周知,同一组分在不同紫外波长下表现出不同的吸收特性。由于退热解毒注射液成分复杂,主要包括有机酸类、黄酮类、酚类、萜类等。样品中活性成分绿原酸的最大吸收波长在330 nm左右;柴胡皂苷D在254 nm有最大吸收峰;丹皮酚、连翘酯苷A、芦丁、木犀草苷等分别在274、330、260、360 nm处有吸收。显然,单一波长表达的信息量有限,为了获得更全面和更真实的样本信息,研究采用DAD检测器记录190~400 nm的紫外光谱(图2),并根据图谱选取化学成分紫外吸收光谱特征较强的5个(210、254、265、330、360 nm)波长,通过融合策略将每个样本在5个检测波长下的积分信号融合成一个统一的色谱指纹图谱。融合方法的原理与三维光谱沿波长轴的投影原理一致(图2)。具体步骤为:整个色谱图数据包含融合基线和融合峰两部分;首先,5个波长的基线数据最大值形成一个新基线;然后根据融合基线上的保留时间对色谱峰排序,将匹配组内不同波长的最大峰指定为融合峰。融合过程采用孙国祥课题组开发的“中药色谱指纹图谱超信息特征数字化评价系统4.0”软件执行,当融合时窗为0.2 min时,有效峰数目与总峰数比值达到最高且每个峰均具有很高的匹配度,与其他时间窗相比,可减少杂峰的影响,充分保留有效峰信息。
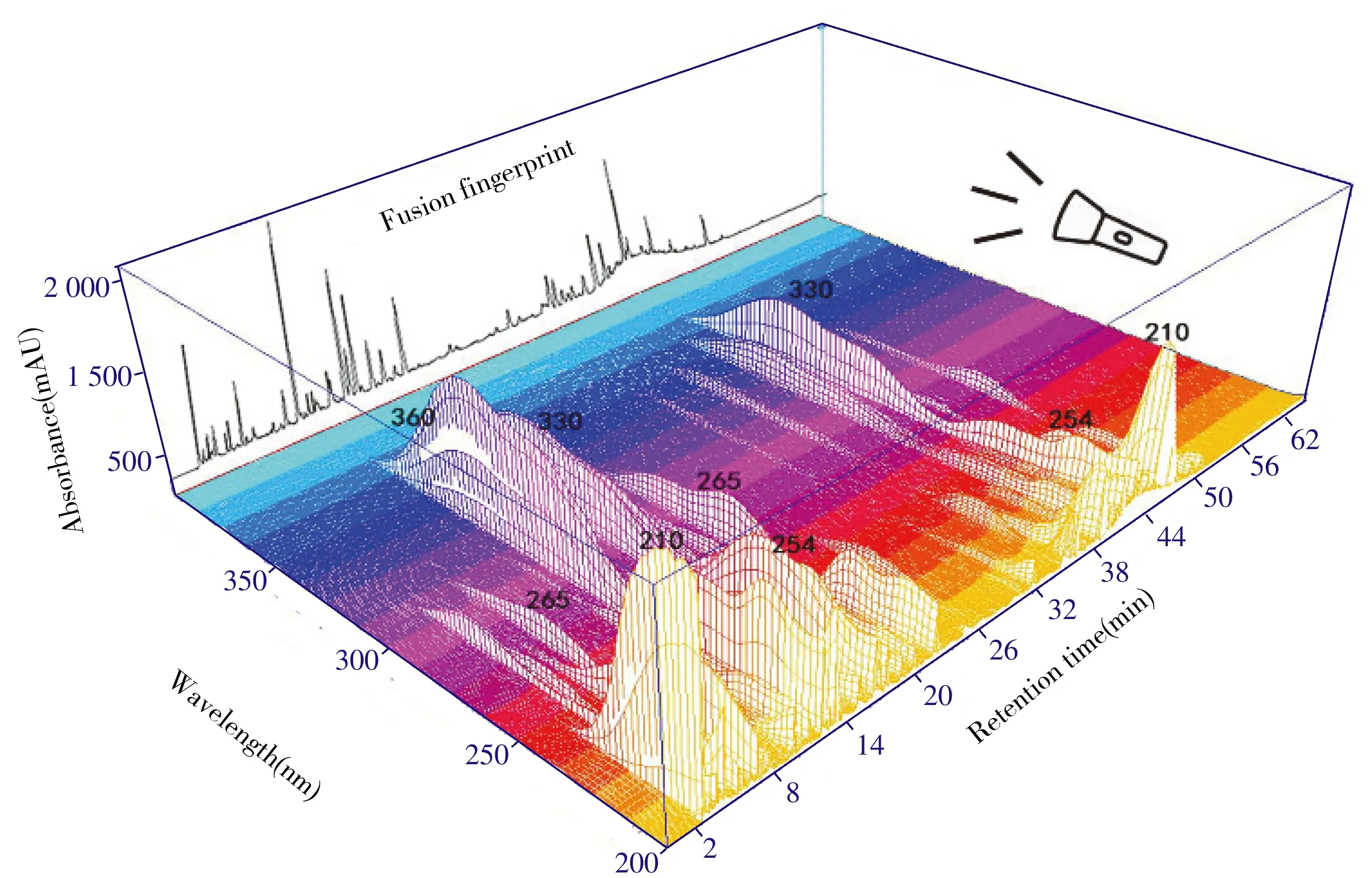
图2 多波长融合指纹图谱原理示意图Fig.2 Principle of multi-wavelength fusion fingerprint(min×nm×absorbance)

图3 20批样品的HPLC融合指纹图谱Fig.3 HPLC multi-wavelength fusion fingerprints of 20 batches of sampes
2.1.3 五波长融合指纹图谱评价利用综合线性量化指纹法对20批样品的五波长融合指纹图谱(图3)进行评价,参考指纹图谱由20批样品的平均值产生。结果显示,20批样品的线性定性相似度Sl值均高于0.970,α值均低于0.1,表明所有样品中化学成分的数量和比例相近。从线性定量相似度Pl%数据可见,不同批次样品间具有显著性差异,有利于将所有样本划分为不同等级(表2)。10批注射液样品被分为5个等级,其中S8质量中等(5级),S5质量良好(4级),S9质量好(3级),其余样品质量为很好及以上(1级和2级)。10批中间体样品被划分成4个等级,其中S12、S14、S18、S20为4级,S11和S15为3级,其余样品划分成1级和2级。S8由于化学成分总体含量显著低于参考指纹图谱(Pl%=70.8)而被评价为5级。不同批次样品的评价等级不同,可能是由于生产工艺或者原料药质量差异所致。另外,10批注射液样品的平均定量相似度为91.0%,10批中间体的平均定量相似度为106.8%,可见从中间体制成注射液的工艺过程中,整体化学成分的含量有所下降,据此可进一步对其工艺过程进行调整或从原料药源头进行质量控制,从而提升样品质量水平。综上所述,综合线性量化指纹法是全面监控不同批次样品质量的有力工具。

表2 综合线性指纹法评价20批样品的质量结果Table 2 Comprehensive linear quantitative fingerprinting method to evaluate the quality and IC50 values of 20 batches of samples
2.2 指标成分含量测定
2.2.1 方法学验证取绿原酸、连翘酯苷A、丹皮酚、柴胡皂苷D对照品适量,分别用甲醇配成6个不同系列浓度对照品溶液,平行测定2次,以对照品浓度(x)为横坐标,对应平均峰面积(y)为纵坐标,绘制标准曲线,再以10倍信噪比(S/N=10)计算定量下限(LOQ),以S/N=3计算检出限(LOD)。另外,平行制备6份已知含量的供试品溶液,分别向其中加入各对照品溶液适量,按“1.3”方法处理并测定,计算各成分回收率。结果显示,4个指标成分在一定质量浓度范围内线性良好,相关系数(r)均不低于0.999 9,LOD和LOQ分别为0.18~10.03、0.58~30.08 μg/mL,平均回收率分别为100%、98.1%、98.6%、97.9%(表3)。由此可见,本方法的准确度良好,对高效液相色谱定量分析具有足够的可靠性和准确度。
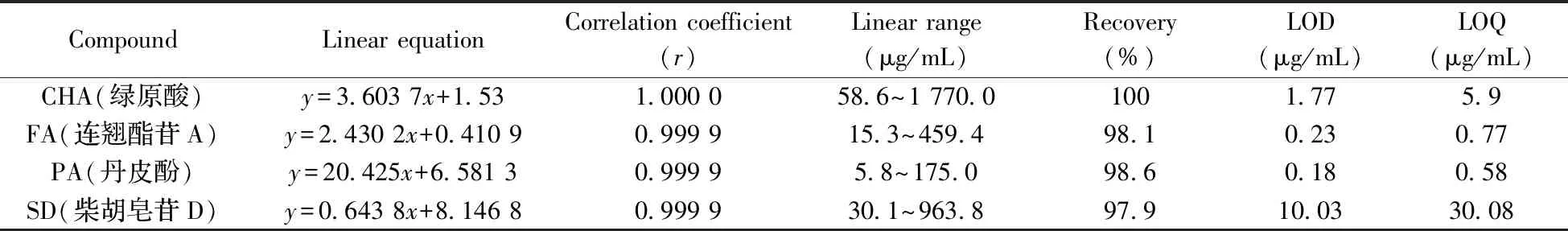
表3 指标化合物的线性方程、相关系数(r)、线性范围、检出限和定量下限结果Table 3 Linear equations,correlation coefficients,linear ranges,recoveries,LODs and LOQs of 6 markers compounds
2.2.2 指标成分定量分析对20批样品采用本方法在优化条件下进行定量分析(表4),得绿原酸、连翘酯苷A、丹皮酚、柴胡皂苷D的平均含量分别为762.00、249.89、74.73、321.54 μg/mL,其中绿原酸的平均含量最高,丹皮酚的平均含量最低。不同批次样品4个指标成分的总含量有明显差异(RSD>12%),这可能与原料药产地、采收季节、制剂生产工艺等差异因素有关。其中S8总含量明显低于其他样品,S20的总含量高于其他样品,并在指纹图谱评价过程中发现S8的Pl%最低,而S20的Pl%最高,由此可见指标成分定量结果与指纹图谱的定量评价结果相一致。此外,当比较同一批次注射液和中间体样品时(比如S1和S11,S2和S12等),可发现中间体的六指标成分含量总和高于对应的注射液样品,表明在中间体制成注射液的工艺过程中,指标成分含量有所下降。

表4 4个成分的定量结果Table 4 Quantitative results of 4 components ρ/(μg·mL-1)

(续表4)
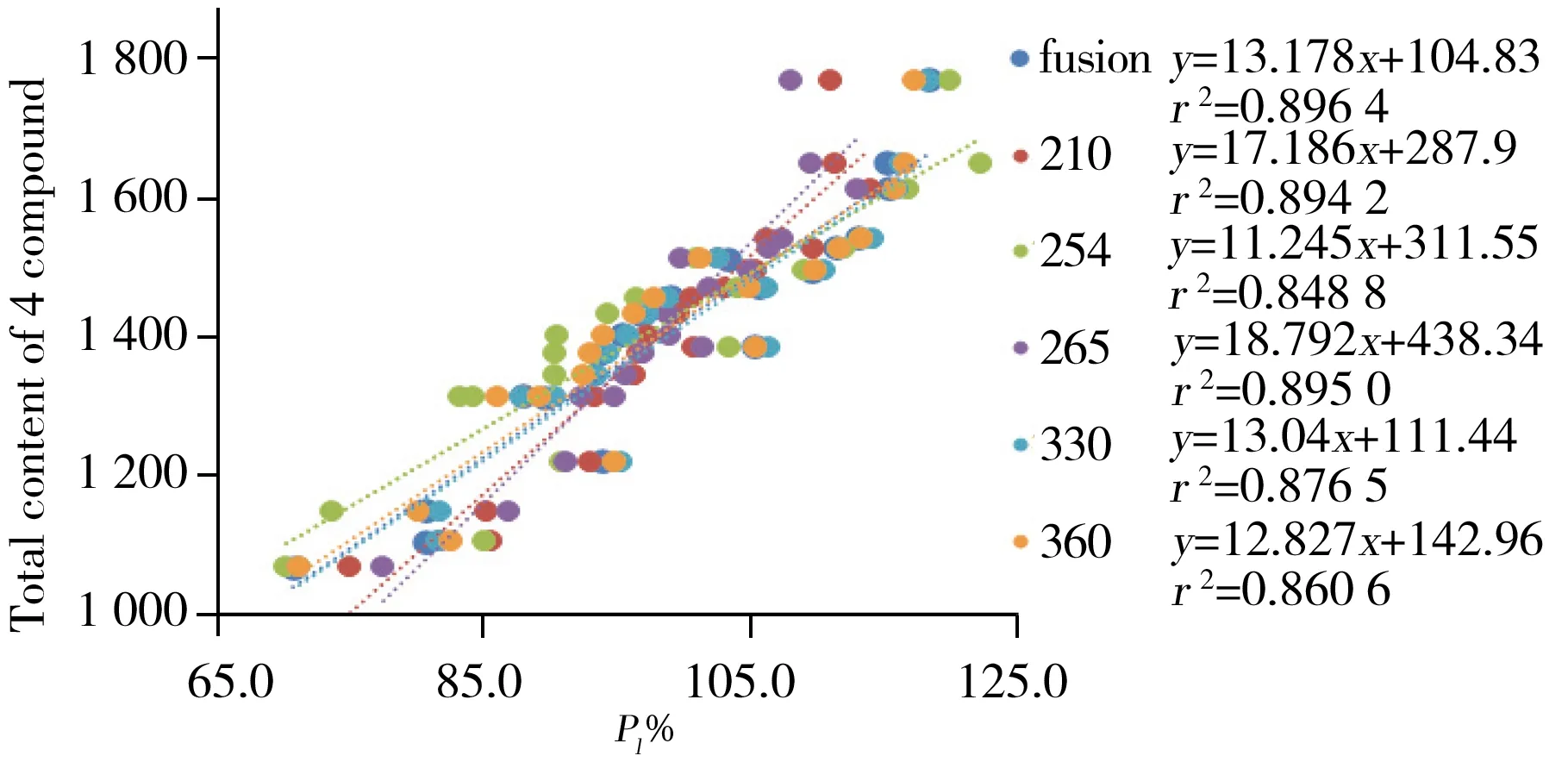
图4 指标定量结果与五波长下指纹图谱定量结果 的相关性研究Fig.4 Correlation study between index quantitative results and fingerprint quantitative
2.2.3 指标成分定量结果与指纹图谱定量参数的相关性由20批样品的指纹图谱评价结果可知,样品质量等级差异主要来源于Pl%,说明Pl%在指纹图谱评价中起重要作用。为进一步探索指标成分定量结果和指纹图谱定量评价之间的关系,实验以每个样本4个成分的含量总和P4c为纵坐标,以不同波长下的定量相似度(Pl%-fusion,Pl%-210 nm,Pl%-254 nm,Pl%-265 nm,Pl%-330 nm,Pl%-360 nm)为横坐标进行线性回归(图4),得到的相关系数(r2)均高于0.84,特别是Pl-fusion和P4c之间的相关系数达到0.896 4,这表明指纹图谱的定量评价与多组分的定量分析间具有较好的相关性。对于大多数中药来说,标准物质的缺乏常使其定量分析变得困难,而综合线性量化指纹法能够全面反映样品中的共有指纹峰含量信息,是一种简便、快速的潜在替代方法。
3 结 论
本文基于退热解毒注射液样品不同化学成分的紫外吸收特征,选取具有特征性和互补性的5个波长(210、254、265、330、360 nm)下的色谱图进行数据融合,得到的融合指纹图谱能够避免单一波长下信息的缺失,更加全面和真实地反映样品质量。并首次提出了综合线性指纹图谱评价方法,该方法科学地提供了综合线性定性相似度(Sl)、综合线性定量相似度(Pl%)和指纹变异系数(α)3个评价参数,并成功地将其应用于样品指纹图谱的整体定性和定量评价。所有样品均显示相似的Sl(>0.970)和α(<0.1);但Pl%展现了明显的变化,说明不同批次样品之间整体化学成分的含量具有一定差异。此外,Pl%与所研究的4种指标化合物的含量高度相关,说明综合线性指纹法可替代指标成分用于复杂系统的定量分析。与传统的相似度方法比较,综合线性指纹法对于中药的质量控制更加严格,有助于规范中药质量生产,提高质量标准。该方法也可作为原料药投料生产、制剂工艺过程中的质量监控手段,为生产实践和临床治疗提供有力的参考和指导。但由于条件限制,本研究仅选择了具有代表性的5个波长,为了更加充分地提取样品三维原始数据信息,进一步的光谱色谱整体分析是需要的。随着计算机的不断发展,数据处理方法的不断完善,全波长范围DAD数据融合技术对于中药质量分析将更具有意义。本研究中五波长的融合虽然不能代表所有样品原始信息,但可以展现不同批次间样品的质量差异,为将来的研究工作提供初步探索。
