基于密度泛函理论的并四苯分子结构与振动光谱研究
2021-01-27范青杰赖仕全朱亚明赵雪飞
范青杰,赖仕全,岳 莉,朱亚明,赵雪飞
(辽宁科技大学 化学工程学院,辽宁 鞍山 114051)
并四苯(C18H12)又称2,3-苯并蒽,是一种线性渺位缩合的多环芳烃(PAHs),由四个苯环线性链接而成,其价态电子结构中包含有大型π共轭电子体系[1]。作为一类重要的有机半导体光电材料,并四苯及其衍生物在发光二极管、场效应晶体管和碳量子点等领域有良好的应用前景[2-4]。与从煤焦油或石油渣油重质馏分中分离获得的萘、蒽、菲等小型线性并苯不同,并四苯以上的大型线性并苯在自然资源中很少存在,常需要通过多步反应合成才能得到[5-7],但由于在自然环境条件下这些大型并苯的高化学反应活性,使得它们的合成具有非常高的难度,进而导致长久以来人们对大型线性并苯的分子结构、理化性质等认识不够深入。目前,有关并四苯分子的结构、分子轨道、分子振动等微观性质的研究尚较少[8]。
近年来,用量子化学方法来研究分子的微观性质受到人们的关注,其中研究多电子体系电子结构的密度泛函理论(DFT)是最常用的较好方法之一[9]。通过对分子进行DFT计算,可得到优化的分子结构,在此基础上获得分子的红外光谱、拉曼光谱以及力学常数和热力学常数等微观特性。本文基于密度泛函理论,研究了线性并苯中并四苯的分子结构和振动光谱特征,以期揭示其电子结构和光谱性质间的关系。同时,通过分析并四苯的前线分子轨道、分子静电势及Mulliken 布居,获得它的HOMO-LUMO能隙、分子静电势图和原子电荷分布等微观性质,为其它大型线性并苯的光谱检测及其光谱和电子结构、反应性特征的分析提供理论基础。
1 理论计算
先构建并四苯的分子模型,再利用密度泛函理论,在B3LYP/6-311++G(d,p)水平下采用Gaussian 09W 程序包对并四苯分子进行结构优化和振动频率计算,获得未矫正的振动频率、红外光谱、拉曼光谱、静态极化率和热力学常数等,最后借助于Gauss View 5.0程序完成分子的前线分子轨道、分子静电势、Mulliken布居和振动归属分析。由于理论计算的振动频率存在系统误差,因此要乘以相应的矫正因子,并四苯的矫正因子为0.98[9-10]。
2 结果与讨论
2.1 分子结构优化
对并四苯分子进行结构优化,计算获得的所有振动频率均为正值,且无虚频,表明优化后的分子结构为稳定构象。优化后的并四苯分子结构及原子编号如图1 所示。并四苯分子优化后的全局最小能为-693.329 361 45 a.u.。为了方便后续分子振动频率的归属,将图1中的苯环从左至右编号为1、2、3、4。
表1 列出了优化后并四苯分子中各原子间的键长、键角和二面角等结构参数,括号中的数字是原子编号,R是键长(nm),A是键角(°),D是二面角(°)。并四苯分子有33 个键(包括12 个C—H和21个C—C)、54个键角和84个二面角。并四苯的 C—H 键长约为 0.108 5 nm,其 C—C 键长在0.136 0~0.145 0 nm 范围,因周围环境而异。并四苯的键角在120°左右,二面角为0°或180°。并四苯是具有高度对称性的平面线性分子。

表1 B3LYP/6-311++G(d,p)水平下优化后的并四苯结构参数Tab.1 Structural parameters of tetracene optimized at B3LYP/6-311++G(d,p)level
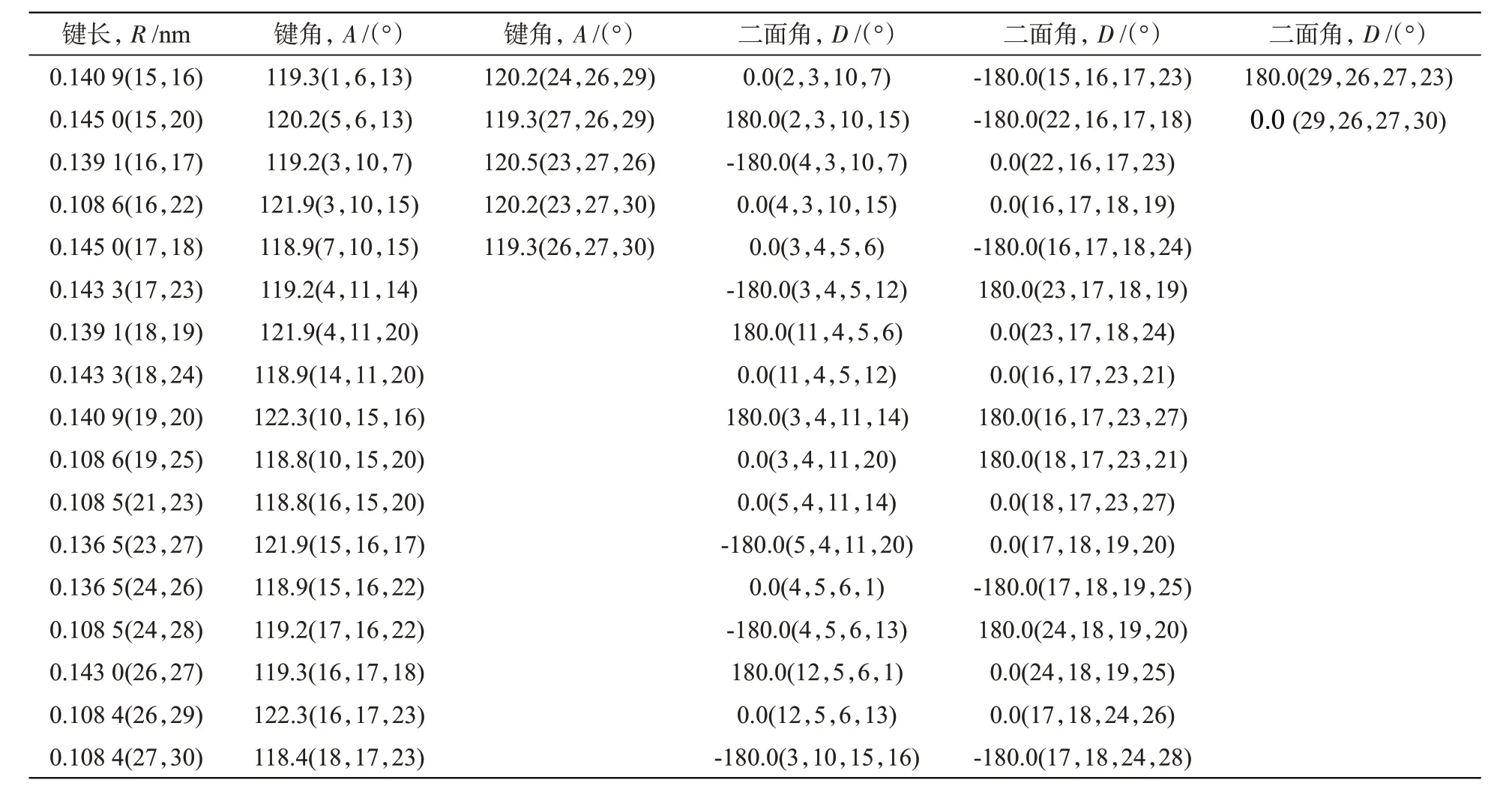
续表1 B3LYP/6-311++G(d,p)水平下优化后的并四苯结构参数Tab.1 Structural parameters of tetracene optimized at B3LYP/6-311++G(d,p)level
2.2 振动光谱
2.2.1 振动频率计算 在B3LYP/6-311++G(d,p)水平对优化后的并四苯分子进行振动频率计算,结果表明:并四苯分子属于D2h点群,由18个碳原子和12 个氢原子组成,包括84 种振动模式,归属后的对称种类为
这84种振动模式包括57种面内对称振动(inphase,A′)、27 种面外对称振动(out-of-phase,A″)。同时,有39个振动为红外活性振动,51个振动为拉曼活性振动。表2 列出了并四苯分子的这84 种振动模式的对称要素、矫正及未矫正的计算频率、实测频率、红外强度、拉曼活性和振动归属。表2 中的红外光谱和拉曼光谱的实测频率数据来自SDSB图谱库及文献[8,11]。
2.2.2 IR 和 Raman 光谱 在 B3LYP/6-311++G(d,p)水平计算获得的红外光谱和拉曼光谱如图2所示。在红外光谱中,并四苯分子有13 个较为明显的红外振动峰,分别位于3 188、1 674、1 581、1 430、1 367、1 314、1 149、1 020、979、922、759、562、474 cm-1处。与文献[11]和SDSB图谱数据库中压片法获得并四苯的红外光谱相比,虽然峰强
度存在一些差异,但峰位置基本相同,尤其是矫正后的峰位置具有良好的一致性。在拉曼光谱中,并四苯分子有8个较为明显的拉曼特征峰,分别位于762、1 021、1 224、1 423、1 478、1 575、1 646、3 164 cm-1处,与文献[8]报道的并四苯实测的拉曼光谱的出峰位置有较好的一致性。

表2 并四苯的计算振动频率及归属Tab.2 Calculated vibrational frequencies and assignments of tetracene
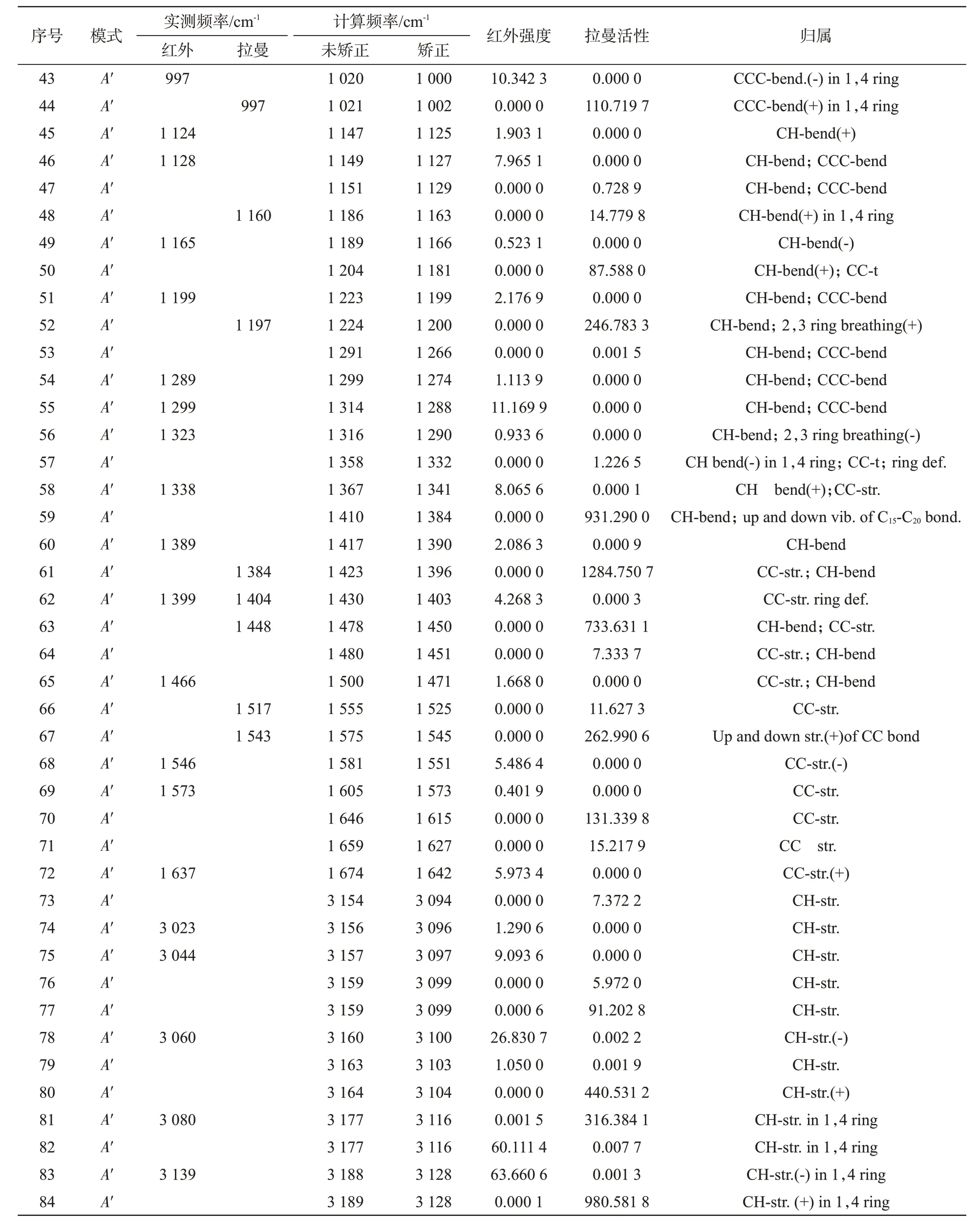
续表2 并四苯的计算振动频率及归属Tab.2 Calculated vibrational frequencies and assignments of tetracene
2.2.3 振动归属 根据图2中的光谱特征,将并四苯分子的振动光谱从高频区到低频区划分为5个区(有些区部分重叠):
(1)3 200~3 000 cm-1区:线性并苯是由多个苯环线性链接而成,它们在结构上与苯相似,而苯环在3 000~3 010 cm-1的吸收峰归因于C—H 伸缩振动。参照SDBS图谱库中并四苯的红外谱图,同时根据Gaussian计算结果,可判断在该区域的3 139、3 080、3 060、3 044、3 023 cm-1处的红外吸收峰源于并四苯的C—H伸缩振动。
(2)1 700~1 200 cm-1区:该区域的振动情况较复杂,存在多种振动模式重叠现象,既有苯环的C—C 伸缩振动,也有苯环的C—H 面内弯曲振动以及苯环的骨架振动或变形。结合实测光谱和Gaussian计算结果,可判断在该区域的1 641、1 573、1 546、1 466、1 399、1 338、1 299 cm-1处的红外吸收峰和1 543、1 517、1 448、1 404、1 384 cm-1处的拉曼特征峰都主要源于并四苯的C—C伸缩振动。
(3)1 100~1 400 cm-1区:该区域的振动也较复杂,主要为苯环的C—H 面内弯曲振动,同时存在苯环的C—C 扭转振动、苯环呼吸振动、C—C—C面内弯曲振动等模式。结合实验光谱和Gaussian计算结果,可判断在该区域的1 399、1 338、1 299、1 199、1 165、1 128、1 124 cm-1处的红外光谱和851、752 cm-1处的拉曼光谱主要源于并四苯的C—H面内弯曲振动。
(4)1 000~700 cm-1区:该区域的振动主要为苯环的C—H 面外弯曲振动。可以判断在该区域的965、956、933、895、743、741 cm-1处的红外峰和1 197、1 160 cm-1处的拉曼峰均源于并四苯的C—H面外弯曲振动。
(5)700~0 cm-1区:该区域的振动模式主要包括苯环的C—C 扭转振动以及苯环的C—C—C 对称或不对称弯曲振动。而苯环的C—C—C对称或不对称弯曲振动在该区域内通常成对出现,如552 cm-1处的红外峰和618 cm-1拉曼峰分别源自图1中1、4 苯环的C—C—C 不对称和对称弯曲振动,628 cm-1处的红外峰和494 cm-1拉曼峰则分别源自并四苯四个苯环的C—C—C 不对称和对称弯曲振动。在小于300 cm-1区域的光谱峰主要归因于苯环的碟形振动、折叠振动以及扭转振动。
2.3 前线分子轨道分析
前线分子轨道是指分子吸收能量后电子从最高占据轨道(HOMO)跃迁到最低空轨道(LUMO)或更高能级的轨道,作为量子化学的一个重要参数,其对分子理化性质的研究十分重要。同时,分子的前线电子云密度可用于预测分子π键中最高活性位置以及共轭体系中的反应类型[12]。HOMO代表提供电子的能力,而LUMO 代表接受电子的能力,二者的能量差被称为HOMO-LUMO 能隙,是电子激发过程所需的最低能量。能隙是反映物质导电性和发光性的一个重要物理量,通常共轭分子的能隙都较小。
图3 给出了并四苯分子的前线分子轨道图和计算的轨道能(EHOMO和ELUMO)。并四苯的EHOMO和ELUMO分别为-5.20 eV 和-2.448 eV,其能隙为2.752 eV。并四苯分子具有较低的HOMO 能,表明其提供电子能力较弱;LUMO能也较低,表明其接受电子能力较强,更容易受到亲核试剂的攻击。并四苯分子含有离域共轭大π键,其HOMO→LUMO的过程实质就是π*→π电子的跃迁过程。
2.4 分子静电势分析
分子静电势(MEP)可反映分子间相互作用的情况,其作为分子微观性质的一个重要物理量,可用于研究分子的反应活性、反应位点、反应类型以及进行分子识别。图4 给出了并四苯分子结构优化后的表面静电势3D图。在MEP图中,深色代表负电势区域,与亲电反应相关,浅色代表正电势区域,与亲核反应相关。并四苯分子中C 原子附近为负电势区域,在其上方形成离域的共轭大π键。相对中间C原子而言,左右两侧的C原子的负电势更大,表明其更易引起亲电试剂的攻击,发生加成反应。并四苯分子中H 原子附近为正电势区域,相对左右两侧苯环的H原子而言,中间两苯环的H原子具有更高的正电势,表明这些位置更易受到亲核试剂的攻击,发生取代反应。
2.5 极化率分析
分子极化率是分子感应偶极矩与电场强度的比值,反映了在电场中分子的极化程度,是研究分子光学性质的一个重要参数。本文利用B3LYP/6-311++G(d,p)计算了并四苯分子在各个方向的精确静态极化率(α)和估计静态极化率(α′),数据列于表3中。根据平均静态极化率计算公式
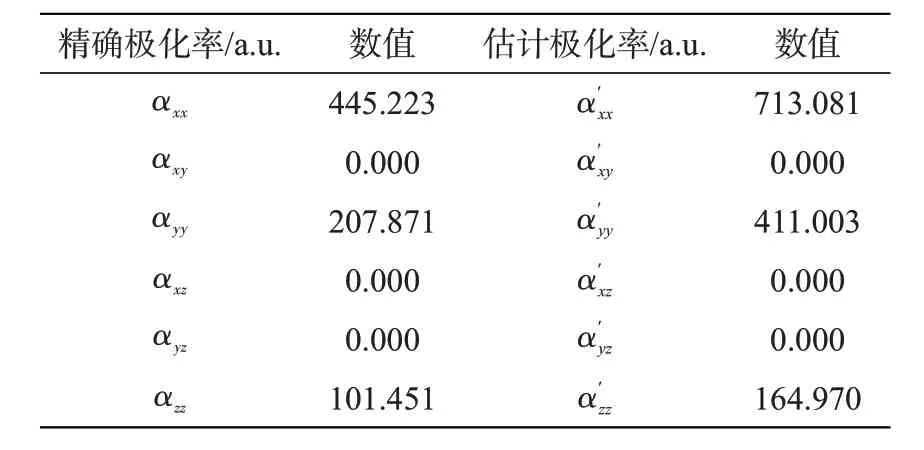
表3 B3LYP/6-311++G(d,p)计算的并四苯分子极化率Tab.3 Polarizability of tetracene calculated at B3LYP/6-311++G(d,p)level
2.6 Mulliken布居分析
通过Mulliken 布居分析可得到分子中各原子的电荷,这有助于了解分子的极化率、偶极矩、电子结构等微观性质[12]。表4 给出了并四苯分子中各C、H 原子所带电荷的情况。在并四苯分子中,12 个与H 相连的C 均处于负电荷区域,且位于左右两侧苯环中的C1、C6、C26、C27 的电负性最大,电荷均为-0.311。在中心位置的C3、C4、C15、C17、C18、C20 六个非质子C 均处于正电荷区域,其中C15 和C20 呈现出最强的电正性,电荷为0.308。另外,C2、C3、C10、C15、C16、C23 明显地存在正、负电荷交替分布的情况,表明四个苯环形成了共轭体系。位于四周的12个H原子均处于正电荷区域,其中位于中间苯环上的H7、H14、H22、H25 具有最小的正电性,电荷为0.083。总之,并四苯分子结构的高度对称性导致其内部C、H原子所带电荷也呈对称性出现。
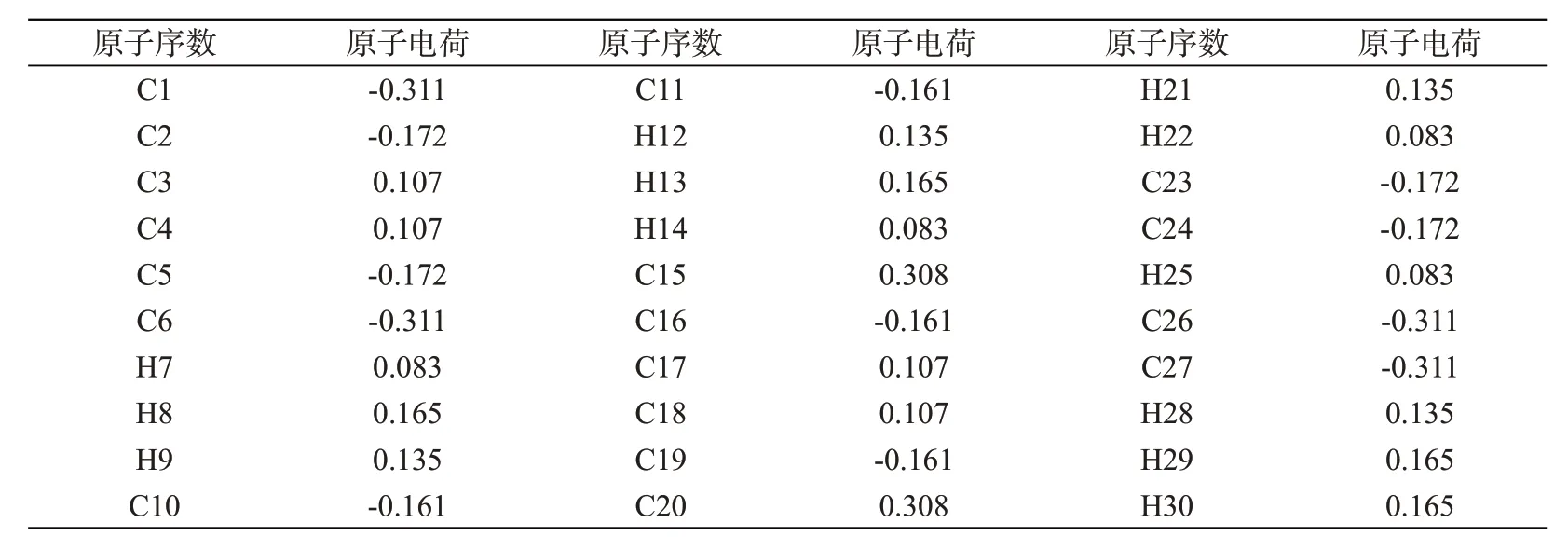
表4 B3LYP/6-311++G(d,p)水平计算的并四苯分子Mulliken原子电荷Tab.4 Mulliken atomic charges of tetracene calculated at B3LYP/6-311++G(d,p)level
2.7 热力学参数分析
在B3LYP/6-311++G(d,p)水平计算并四苯分子的零点振动能Ezpv=628.698 kJ/mol,在x、y、z方向上的转动常数CR分别为1.637 97、0.213 86、0.189 16 GHz,对应转动温度TR分别为0.078 61、0.010 26、0.009 08 K。表5还列出了并四苯分子在平动、转动、振动时的能量E、恒容热容CV、熵S等热力学参数。
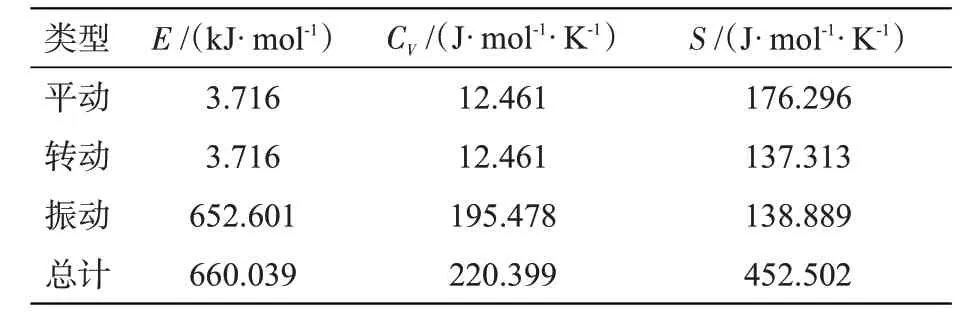
表5 B3LYP/6-311++G(d,p)水平计算并四苯分子的热力学参数Tab.5 Thermodynamic parameters of tetracene calculated at B3LYP/6-311++G(d,p)level
3 结 论
利用密度泛函理论在B3LYP/6-311++G(d,p)水平得到了并四苯的分子结构参数、红外光谱和拉曼光谱。结果表明,并四苯的计算频率与实测频率之间有良好的一致性,其分子结构的高度对称性导致其表面静电势和原子电荷分布也呈一致的对称性。并四苯分子具有一定的非线性光学特性,其HOMO-LUMO能隙为2.752 eV,中间苯环上的H 易被取代,两侧苯环易发生加成反应。本文研究为并四苯的光学性质及其化学反应性提供重要依据,同时也为其它大型线性并苯的分子结构与振动光谱的理论计算和微观性质分析提供参考。
