基于拉普拉斯规范化和重启随机游走算法的微生物疾病关系预测
2021-01-26罗洪,杨杰
罗 洪,杨 杰
(西南民族大学计算机科学与技术学院,计算机系统国家民委重点实验室,四川 成都 610041)
人类是细菌、病毒和真菌等微生物的宿主,这些微生物通常存在于人体的肺、皮肤、肠道和口腔等人体器官中.大多数微生物具有促进人体新陈代谢的能力以及协助调节胃肠道的发育的功能.据报道,在一个成年肠道中生活着大量的微生物,它们可以提供多种基因产物,这些基因产物有助于人体的各种生化和代谢活动[1-3].例如常见的肠道细菌可以通过帮助消化多糖来促进营养的吸收.反之,微生物群落异常水平的失衡会影响人类的健康和疾病的发生,甚至影响基因的转录和表达,从而导致癌症、糖尿病、肥胖症和过敏性哮喘等疾病的发生[4-9].
研究潜在的微生物疾病间的关系可以帮助了解疾病的发病机理,为有关疾病的预防、诊断和治疗提供帮助.传统生物实验方法存在研究周期长成本昂贵等问题,而采用计算方法有助于传统方法确定研究目标范围和缩短研究周期.近年来,国内外学者运用多种计算方法已经对疾病相关的微生物进行了预测研究.例如文献[10]基于KATZ指标首次提出了KATZHMDA计算模型对潜在微生物和疾病的关系进行了预测;文献[11]采用基于路径的方法提出了PBHMDA算法对潜在微生物和疾病的关系进行了预测;文献[12]基于双随机游走提出了BiRWHMDA计算方法对潜在微生物和疾病的关系进行了预测.本文利用已知微生物疾病关系相互作用网络、拉普拉斯规范化后的高斯核微生物和疾病相似性网络构建,构建了异质网络,并利用重启的随机游走算法在异质网络中游走预测潜在的微生物和疾病的关系.本文算法LRWRHMDA经过在拉普拉斯规范化后的高斯核微生物和疾病相似性网络构建的同一异质网络数据集上与KATZHMDA和BiRWHMDA算法进行五折交叉验证比较,实验结果验证了本文算法LRWRHMDA的有效性.
1 算法描述
1.1 拉普拉斯规范化
若矩阵L=[L(i,j)],i,j=1,2,3,…,N,是个对称矩阵.D是矩阵L的对角矩阵:当i≠j,D(i,j)=0;当i的值等于矩阵L第i行的和.矩阵L通过规范化,结果仍然是一个对称矩阵.对称矩阵的元素可表示为:

上述过程就是矩阵L的拉普拉斯规范化过程,可用于网络带权矩阵的规范化,尤其是随机游走算法中概率转移矩阵的规范化[13].
1.2 微生物和疾病高斯核相似矩阵及其规范化
已有研究表明存在这样的假设:功能相似的微生物常常与相同的疾病相互作用.根据这样的假设我们可以利用已知的微生物和疾病相似性网络计算微生物和疾病各自高斯相似性矩阵.已知微生物和疾病网络的邻接矩阵AMD,对于微生物mi,存在行向量Mmi,若值为0表示和已知某种疾病无关,若为1表示和某已知疾病相关.则对微生物mi和mj间的高斯核相似性GSM可表示为:

其中参数γm控制核的范围,它可通过另一个范围参数γ′m通过每个微生物与疾病的平均关联数规范化后求得.参数γm的计算公式如下:

同理,疾病di和dj间的高斯核相似性GSD可由以下公式求得:

1.3 异质网络上的重启随机游走
若存在已知的微生物和疾病关系网络的邻接矩阵AMD、疾病di和dj间的高斯核相似性GSD和微生物mi和mj间的高斯核相似性GSM,则异质网络H的邻接矩阵可表示为:

其中,AMD、LGSm(m×m)和LGSD(n×n)分别代表已知微生物和疾病邻接矩阵、拉普拉斯规范化后的微生物和疾病高斯核相似矩阵,AMDT表示矩阵AMD的转置.
异质网络H的概率转移矩阵可以表示为:其中MGSM和MGSD表示微生物和疾病子网的概率转移矩阵,MMD和MDM表示网间概率转移矩阵[14].
令λ表示网间的跳转概率,则从微生物mi跳转到疾病dj的转移概率可表示为:
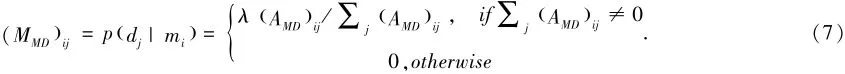
同理,从疾病dj跳转到微生物mi的转移概率可表示为:

从微生物mi跳转到mj的概率可表示为:

从疾病di跳转到dj的概率可表示为:
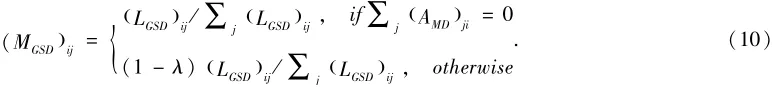
令微生物网络的初始概率为μ0,疾病网络的初始概率为v0.则异质网络H的初始概率p0可表示为:

令γ∈(0,1)表示重启概率,M表示异质网络的转移矩阵,则第t+1步的概率pt+1可表示为:

每游走一步,随机游走者返回种子点的概率都为γ,当随机游走到一定步后,概率将达到一个稳定状态,此时微生物和疾病则可基于各自的稳态概率来排序,从而预测微生物和疾病间的关联关系.
2 实验分析
本文采用HMDAD数据库[15]中的数据集进行LRWRHMDA算法的致病微生物预测应用分析.该数据集从已发布的文献中收集和整理了39种人类疾病、292种微生物和483对已知微生物-疾病关系对.经过整理去掉重复的关系对,本文以39种人类疾病、292种微生物和450对已知微生物-疾病关系对为基础构建微生物和疾病网络邻接矩阵.
交叉验证的基本思想把数据集划分成较小子集的训练集合测试集,训练集用于模型的训练,测试集用于验证模型的稳定性和可靠性.K-折交叉验证通常把数据集分成k份,一份用作测试集,其它K-1份用作训练集,每次验证一份重复K次,结果取K次的平均值.本文采用5-折交叉验证类评估LRWRMDH算法的性能,其中5-折交叉验证重复执行100次再取平均值.并用经过拉普拉斯规范化后的高斯核微生物和疾病相似性网络构建的同一异质网络与KATZHMDA和BiRWHMDA算法进行了5-折交叉验证比较,5-折交叉验证下LRWRHMDA、BiRWHMDA和KATZHMDA的AUC值分别为0.9069、0.8844和0.8127,得到的ROC曲线[16]如图1所示.
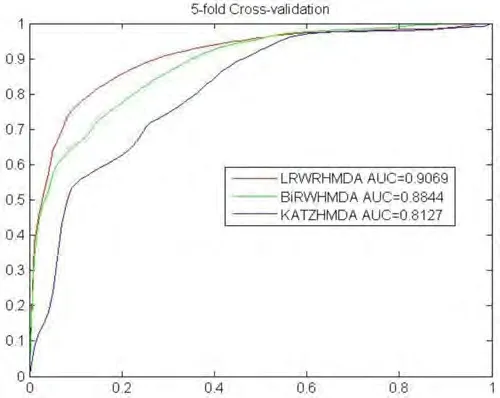
图1 三种方法5-折交叉验证ROC曲线图Fig.1 The ROC curves of three methods based on 5-fold CV
为了进一步验证本文算法的预测效果,我们对哮喘(Asthma)和炎症性肠病(IBD)预测的相关排名前10位的微生物进行了验证,结果如表1和表2所示.

表1 预测哮喘相关前10位微生物Table 1 Prediction results of the top 10 asthma-associated microbes

表2 预测炎症性肠病相关前10位微生物Table 2 Prediction results of the top 10 IBD-associated microbes

4 Veillonella unconfirmed 5 Haemophilus unconfirmed 6 Clostridiumcoccoides PMID:19235886 7 Bacteroidaceae Maukonen et al.,2009 8 Bacteroides Maukonen et al.,2009 9 Streptococcus PMID:23679203 10 Lactobacillus PMID:26340825
综合实验分析可知,LRWRHMDA与其它两种算法相比五折交叉验证下的AUC值较优,预测的预测哮喘相关前10位微生物仅排在第10位的没有得到已知文献验证,预测炎症性肠病相关前10位微生物仅排在第4位和第5位的没有得到已知文献验证.
3 结论
微生物对人类健康的重要作用得到了越来越多的研究证明,研究微生物与疾病的关联关有助于揭示疾病的发病机理和疾病的诊断和预防.在本文中我们基于已知微生物疾病关系互作网络、拉普拉斯规范化高斯核微生物和疾病相似性网络构建的异质网络上的重启的随机游走算法,对微生物和疾病的潜在关系进行了研究和预测,并与其它预测算法进行了五折交叉验证比较.比较结果和预测验证结果验证了本文算法的有效性.鉴于本文算法采用数据集的单一性和异质网络游走依赖于已知的关系对,因此本文算法也存在局限性,这是将来需要进一步改进的方向.
