微生物组学及其在厌氧消化中的研究进展
2021-01-22李叶青景张牧江皓徐泉周红军冯璐
李叶青 景张牧 江皓 徐泉 周红军 冯璐
(1. 中国石油大学(北京)新能源与材料学院 生物燃气高值利用北京市重点实验室,北京 102249;2. 丹麦奥胡斯大学工程系,切勒 8830)
目前,全国每年产生数以亿计的有机废弃物。现阶段常用的有机废弃物处理方法包括:热处理(如气化、热解等)以及生物处理(如厌氧消化、堆肥等)[1]。我国城市有机废弃物的含水率高、热值低(热值为3 MJ/kg,含水率约80%[2]),导致热处理的效率不高。而堆肥由于产生大量温室气体(CO2、CH4和N2O)、占地面积大以及周期较长等缺点,限制了其直接应用[3]。不过,好氧堆肥可以作为厌氧消化的后续处理使沼渣深度腐熟,转化为腐殖质改良土壤[4]。将厌氧消化和好氧堆肥联合使用,既解决了厌氧消化后产生的沼渣不宜直接还田的问题,还可缩短好氧堆肥周期、提高有机肥肥力。此外,从能源结构、碳减排、城镇化发展等多个角度考虑,沼气工程在中国的发展已经成为了必然趋势[5]。到2020年,中国沼气年利用量已达到440×1010m2[5]。尽管厌氧消化技术在我国发展迅速,但其系统中种群结构等信息尚未被完全了解[6],这在一定程度上阻碍了它们在生物能源生产方面的广泛应用[7]。如果仅依赖于微生物培养的技术,微生物活性和功能的生物因素(如种间关系和生物多样性)等对消化过程产生的影响会被忽视。到目前为止,我们仍在努力将厌氧消化系统中的群落结构和功能与环境等因素联系起来,并期望通过改变微生物群落来优化过程效率和提高稳定性[8]。微生物组学分析能够极大的提高我们对这些复杂系统的理解,被认为是了解厌氧消化系统中的群落结构和构建其与外部环境变化关联性的有力工具。本文系统回顾了厌氧消化过程的微生物学研究,并强调各种生物因素对厌氧消化表现的重要影响。
1 厌氧消化的过程
目前,对于厌氧消化过程的认识普遍基于“三阶段、四种群”的基本原理[10]。“三阶段”依次为水解阶段、产酸阶段和产甲烷阶段。“四种群”分别是水解酸化菌、产氢产乙酸菌、同型产乙酸菌和产甲烷菌。水解阶段一般认为是复杂的有机化合物转化为简单的有机化合物的过程,简单的有机化合物分为两类:产甲烷物质和其他消化产物。产甲烷物质是可以直接被产甲烷菌利用并产生甲烷的底物,主要为乙酸(70%)[11]。其他消化产物则能够被产氢产乙酸菌代谢,转化为乙酸、氢气和二氧化碳。与此同时,同型产乙酸菌将氢气和二氧化碳转化为乙酸(此阶段可逆)。最后,在产甲烷阶段,不同营养型的产甲烷菌将产甲烷物质和H2转化为气态甲烷,这是整个厌氧消化的限速阶段[12]。多种微生物发挥不同的功能,共同组成了厌氧消化系统[13]。厌氧消化的基本原理如图1所示。
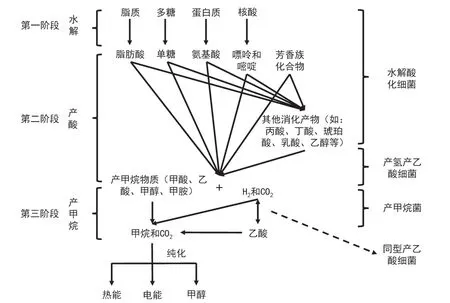
图1 厌氧消化的基本原理(三阶段四种群)
2 微生物组学类型
微生物组学包括基因组学、蛋白组学以及代谢组学,研究微生物从遗传基因到表达代谢的全过程(表1)。基因组学包括16S rRNA基因组学[14]、宏基因组学[15]、宏转录组学[16]。其检测手段为高通量测序技术,又称为下一代测序(Next-Generation Sequencing,NGS)。高通量测序技术至今为止已经经历了三代的发展,第1代高通量测序技术以Sanger测序为代表,第2代以Illumina测序平台为代表,目前正在开发第3代——单分子测序技术[17]。近年来随着测序平台吞吐量的提升、参考数据库的完善以及成本不断下降[18],高通量测序技术已经被广泛用于调查和研究各种厌氧消化系统的生物多样性[19],成为研究各种环境中微生物群落的有力工具。目前最广泛使用的高通量测序系统主要基于二代 Illumina 平台所开发的MiSeq、HiSeq和NovaSeq等[17]。基因组学的研究为微生物群落提供了详尽的物种信息[20]。
总体而言,各种基因测序方法都有其特定的局限性,而利用多种测序方法共同分析可以弥补其不足。以16S rRNA基因测序为例,由于通用引物无法匹配所有16S rRNA靶点,研究的高变区数量有限(大多为V4)以及扩增过程中产生嵌合体等原因,其精度会受到制约,还会使得特定物种的丰度发生改变,例如Euryarchaeota和Spirochaetes[21]。而宏基因组测序则不受系统发育标记基因的限制,能够检测出多种物种(细菌、真菌、古菌、病毒等),并可能获得新基因乃至新物种信息。因此被认为是评估分类学组成最准确的方法[22]。而宏基因组的局限性在于其不能区分表达和非表达的基因,因此不能反映实际的代谢活动[23]。而宏转录组可被用于识别复杂生态系统中mRNA表达的生物学特征[16]。但宏转录组的研究也存在包括从环境样本中难以回收高质量mRNA,物种的mRNA半衰期短,以及需要从其他RNA物种中分离mRNA等问题[22]。因此,从宏基因组分析推断出的基因丰度高并不一定表明具有高的转录或代谢活性。Maus等[24]通过对嗜热沼气工程的微生物组进行研究,发现其中包含大量迄今为止未知或未充分了解的物种,只有18%-25%的16S rRNA基因转录序列能被分类到属级,这表明宏转录组的测序广度还有待提高。截止到目前,被测序和研究的16S rRNA序列,5S和23S rRNA序列较为丰富,mRNA序列则相对较少。尽管宏转录分析的应用中存在需要进一步细化的技术问题,但其已经提供了一些新的生物学发现。Wang等[25]通过宏转录分析和关键酶的测定发现磁铁矿诱导的直接种间电子传递(Direct interspecific electron transfer,DIET)可以部分取代种间氢转移(Interspecies Hydrogen Transfer,IHT),清除电子转移阻断,改善乙酸营养型甲烷生成效率。
宏蛋白质组学被定义为在特定位点的环境微生物群落的所有蛋白质组成进行大规模表征[26],为蛋白质结构、定位和蛋白质-蛋白质相互作用的研究提供最直接的证据。由于不同微生物群落的复杂性、自然环境的异质性和干扰化合物的存在,很难提取出合适的蛋白质部分进行分析[27]。尽管存在这些困难,蛋白质组学在将微生物群落的遗传多样性和活动与其对生态系统功能的影响相关联方面发挥着巨大的潜力[22]。例如,Jing等[28]首次用于混合培养的iTRAQ定量蛋白质组学分析表明,在丙酸盐甲烷化过程中,磁铁矿诱导了涉及各种途径的蛋白表达水平的变化,并发现丙酸代谢相关蛋白上调。现阶段宏蛋白质组学仍是一个新兴领域,其在未来对复杂微生物群落的研究可能得到进一步发展和广泛应用。
近些年代谢组学虽然发展迅速,但是仍然远远落后于基因组学和蛋白质组学,尚未应用于厌氧消化,本文不做讨论。
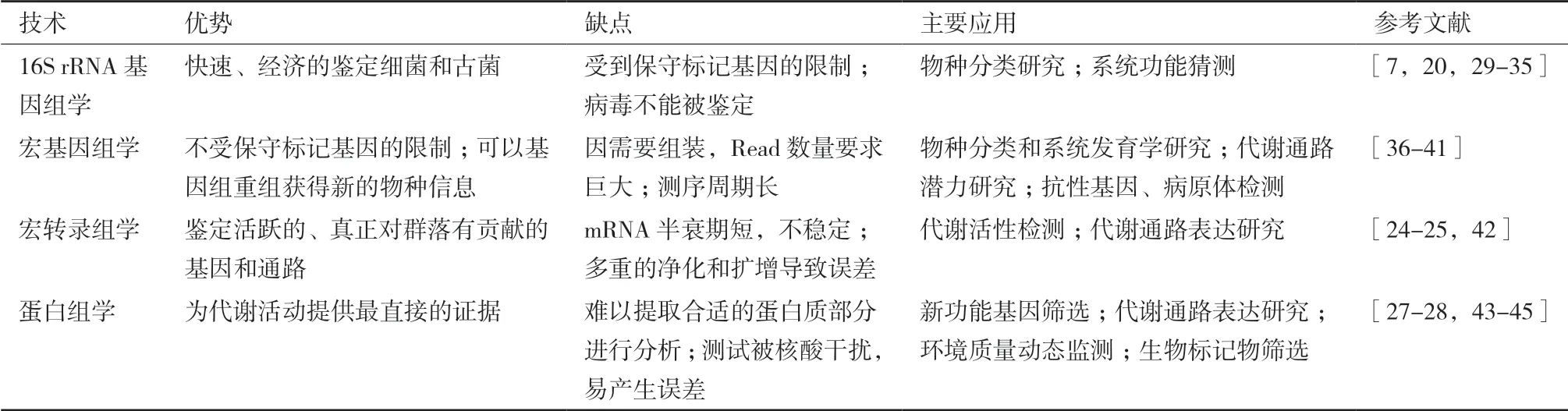
表1 应用于厌氧消化的微生物组学类型
3 常用生物信息学分析手段
常用的厌氧微生物群落生物信息学分析如图2所示。从图中看出,生物信息学分析测序结果包括几个方面,物种组成分析,α多样性分析,OTU聚类分析,多元统计学分析(PCA/ PCoA/ NMD,RDA/CCA),共现关系研究和代谢功能与通路研究。

图2 厌氧微生物群落生物组学分析(部分图片经参考文献[31-32]许可转载,版权归爱斯维尔、美国化学学会所有)
3.1 物种组成分析
物种组成分析是厌氧微生物群落最常用的生物信息学分析方法之一。可以使用Phylophlan[37]、MetaPhlAn[39]、PyNAST[7]或者 RDP Classifier[46]等软件对核心OTU序列和参考序列进行比对,并将比对后的序列进行系统发育组成推断。常用的基因组 参 考 序 列 数 据 库GenBank[46]、EBI、DDBJ[39]、IMG/M[47]。常用的物种注释数据库包括Greengene数据库[31]、Silvia数据库[41,47]。以图2-a为例,微生物群落的系统发育分析可以从界、门、纲、目、科、属、种等不同层次进行,运用Krona软件[40]能够展现出整个微生物群落的多级物种分类图。
目前,学者们对不同操作条件下厌氧消化池微生物组成进行了大规模的分析。其中最常见的研究内容是细菌和古菌的相对丰度。不同底物、接种物以及反应参数下,反应器的微生物组成各不相同[31-32]。但可以确定的是,随着时间的推移,相同反应器微生物丰度在一定时间内的总体概况是相似的,群落演替遵循确定的趋势[31]。
3.2 α多样性分析
通常使用Mothur[20,34]、R语言中的Vegan包[31]或RDP[48]来研究产甲烷系统的微生物群落多样性。常用的多样性分析的指数(图2-b)包括:Chao1丰富估计量,基于覆盖度的物种丰富度(ACE),香农多样性指数(Shannon- Wiener index)和辛普森多样性指数(Simpson diversity index)。其中Chao1、ACE和Shannon- Wiener index与微生物群落生物多样性呈正相关,Simpson diversity index 与微生物群落生物多样性呈负相关。Chao1和ACE指数可直接用于计算微生物群落中物种总数,Shannon-Wiener和Simpson diversity index还与群落均匀度有关,可用于估算生物多样性。群落的均匀度越高,其产甲烷活性就越高。Wittebolle等[49]观察到群落的均匀度与功能稳定性相对应。微生物多样性结构的均匀性确保了群落的多样化代谢的能力。此外,物种的覆盖率(Good’s coverage)越高,测序结果越真实。α多样性分析各指标的计算公式如表2所示。α多样性的下降可作为反应器运行状况恶化的早期预警[47]。

表2 α多样性分析参数的公式和符号意义[50]
3.3 OTU相似性分析
维恩图(VennDiagram)可以用来表示OTU的共同和独特的组成部分[32]。在通常情况下进行分析时,常选用相似水平为97%的OTU 样品。可以使用R 语言VennDiagram包[32]、Venny工具[47]或在线工具(http://www.biovenn.nl/)绘制出图。如图2-c可以直观地反映出厌氧反应器中OTU的相似部分,3个反应器的相同OTU比例为60.9%。结合其他讨论,维恩图的公共部分可能是该系统中发挥特定功能的物种[32]。
3.4 多元统计学分析
最常见的多元分析方法包括主成分分析(Principal Component Analysis,PCA)[43,47],主坐标分 析(Principal Coordinate Analysis,PCoA)[34,51],非度量多维尺度分析(Non-metric Multidimensional Scaling,NMDs)[35,42],冗 余 分 析(Redundancy Analysis,RDA)[7,21,37,48]和典范对应分析(Canonical Correspondence Analysis,CCA)[51]。这些多元技术的共同之处在于,每一种技术本质上都被看作是一种排序分析,其目的是描述相邻对象的相似性。常用的计算工具包括R语言的Vegan包[7]、PC-ORD软件[20]和Matlab[45]等。
PCA与PCoA以及NMDs是3种典型的非约束排序分析。其中PCA与PCoA的一个显著区别在于,PCA基于相似系数矩阵(如欧式距离)确定主成分,而PCoA基于距离矩阵(如Bray-Curtis,UniFrac 和Jaccard)确定主坐标(图2-d)。另一个非约束分类分析NMDS算法能够对对象的距离进行排序,并使用这些排序将对象非线性地映射到一个简化的二维排序空间上,从而保持它们排序的差异而非原始距离[52]。与PCA和PCoA相比,NMDS算法相似度高的数据集更密集,相似度低的数据集更疏远,这使得被测细菌和古菌群落剖面之间关系视图更加清晰[52]。无约束排序分析在分析复杂宏基因组数据,或者微生物群落结构差异方面的应用越来越多[7,31,34]。Feng等[31]运用三维- PCoA分析发现不同污泥停留时间(SRT)的反应器内的微生物群落迅速从接种菌群中发生分化,且微生物群落动态在不同反应器之间同步。Ning等[35]将NMDS结合参数联合解释分析发现,酸和的协同胁迫是抑制机制的主要驱动因素,并进一步发现细菌群落对相关负载蛋白更为敏感,有可能引起产甲烷群落从乙酸营养型产甲烷向氢营养型产甲烷的转变。
在约束排序分析中,RDA和CCA是两种常用的微生物与环境因子关联性分析方法。这两种方法非常相似,都可以使用箭头代表环境因素,解释环境梯度影响了系统发育结构的变化情况。不同的是,RDA基于线性模型,而CCA基于单峰模型。以用Anoca软件进行去趋势对应分析(Detrend Correspondence Analysis,DCA)分析结果作为选择RDA和CCA的标准[20,47]。若DCA第一轴大于4.0,CCA拟合效果更好;而在3.0-4.0之间,RDA和CCA均可;小于3.0,RDA的结果可信度更高。Feng等[31]运用CCA分析表明,随着OLR的增加,碳水化合物、脂类、蛋白质等有机大分子向可溶性有机分子的水解和转化增加。反之丙酸和乙酸等可溶性中间产物浓度的升高可能表示产甲烷过程被部分抑制。
3.5 共现关系研究
厌氧消化菌群并非以单个OTU来执行功能,而是通过厌氧食物链形成一个复杂的群落。为了描述种间关系,共现关系网络常用于揭示物种-物种的关联,描述核心物种之间的相互作用[38,47]。在这些研究中,共现网络的拓扑结构主要在Gephi[31]、Cytoscape[47]软件中使用皮尔逊或斯皮尔曼相关性分析[53]和存在缺失数据的超几何分布[54]得到了可视化。密集连接的节点被划分为簇,这些簇被解释为具有重叠生态位[53-54]的类群。
以图2-e为例,当两个节点(OTUs或任何分类相关的单位)在多个样本中同时出现或显示相似的丰度模式时,则为正相关;反之则为负相关。节点的大小代表与其相关物种的多少,节点之间各边的宽度与相关强度成正比[34]。然而这些关系的生态相关性并不容易解释。例如,由于交叉取食、生物膜内的共聚集、共定殖、生态位重叠等原因,都可能形成正相关关系;而由于偏害共栖、捕食-被捕食关系、竞争等原因,则可能形成负相关关系[55]。此外,更复杂的生态相互作用形式容易被忽视,例如滞后关系和多物种的共生(竞争)关系。
3.6 代谢功能与通路研究
为了深入了解分类组成之外的信息,可以使用生物信息学平台或工具对宏基因组数据进行功能注释。常用读取基因组的工具为MG-RAST服务器[39]。常 用 功 能 注 释 数 据 库 包 括GO、KEGG[36,40,42]、COG[36,41]、Pfam[41]、SWISS-PROT[45]、UniProt-KB[45]。赵一全[42]利用宏转录组学在研究预处理秸秆对微生物的影响时,通过基于KEGG数据库比较甲烷代谢相关途径丰度时发现未处理样本微生物群落能够进行多种途径的营养代谢活动,而经过预处理的样本中的甲烷代谢途径较为单一。之后将注释到的数据映射到代谢通路图(类似于图2-f)中,发现预处理并未改变代谢途径,而是能够将某些通路中的中间代谢产物的合成途径变得更为直接。Heyer等[45]首次对40份沼气厂样本进行了大规模宏蛋白组分析。该方法比起基因组学研究,可以过滤出更具体的核心物种和核心函数。在这些系统研究的基础上,可以确定某种蛋白作为生物标志物。如果这些生物标记物的丰度迅速变化或表现出与选择的工艺参数相矛盾,这可能是之后过程失败的标志。
4 厌氧消化微生物组学的研究进展
4.1 微生物群落与运行参数关联分析
微生物组学分析将微生物群落组合模式与运行参数相联系,可以指导厌氧消化参数的调节。但目前用来解释微生物群落组合模式的理论仍存在一定的分歧。其中占主导地位的理论主要分两种:生态位理论和中性理论。经典的生态位理论强调了微生物的种间关系(如共生和竞争)生态位分化和底物营养等确定性过程的重要性[29]。中性理论则只考虑出生、死亡、漂移、移民等随机过程对生态环境的影响[56]。大部分研究认为生态位理论和中性理论对构建土壤和生物反应器的微生物群落非常重要[57]。微生物群落的组成并不完全是由环境条件决定,而是在一定程度上依赖中性过程[56]。了解微生物群落组成对生态位理论和中性过程及对环境条件的依赖度,对设计和维护厌氧消化工程有着极大的意义。
由于群落组合模式的复杂性以及影响微生物群落结构的参数的多样性,理解环境参数与微生物的相互作用变得困难[58]。一般来说在所有厌氧消化系统中均存在一个核心菌群[37]。Tao等[59]从20个运行了长达两年的实验室规模的厌氧消化反应器中采集了138个样本,并得出了核心菌群的组成(Bacillus、Clostridium、Bacteroides、Eubacterium、Cytophaga、Anaerophaga和Syntrophomonas等),而产甲烷菌的组成因接种源、反应器温度或盐度不同而不同。核心菌群与沼气产量有显著的相关性。产甲烷量高的反应器,其细菌群落非常相似。相对应的是,古菌群落由于功能的变异性较大而具有较强的多样性[37]。此外,有些物种虽然稀有,但也是厌氧消化过程的重要驱动力,如Hyphommicium、Gordonia、Methanospirillum等[48,60]。
4.2 代谢途径分析
最近的研究表明,厌氧消化所涉及的细菌群落具有高度的恢复力(Resilience)。水解、产酸的每一个主要步骤都有多个相关的系统发育群落,功能冗余在所难免[29]。较高水平的发酵菌群(如互营细菌)的动态变化与初级发酵菌群(如Clostridia和Bacteroidetes)丰度变化明显不同,它们更依赖冗余来维持群落的整体功能[37]。均匀度较高的群落利用冗余功能通路的潜力更高,即具有更多并行通路的群落对底物代谢的反应更有效[7]。这使得以特定代谢方式调控细菌菌群变得困难[61]。
作为厌氧消化过程中甲烷生产的最终步骤,产甲烷古菌代谢途径较为单一。目前所有已知的产甲烷菌都表达甲基辅酶M还原酶(mcr)[62]。最常见的乙酸营养型产甲烷菌参与的乙酸代谢过程如图3-A所示:首先,以乙酸受到乙酸激酶(ack)、磷酸乙酰转移酶(pta)或乙酰辅酶A合成酶(acs)等酶催化形成乙酰辅酶A。然后乙酰CoA 再在乙酰辅酶A脱羰酶/合酶复合体(cdh)的作用下形成甲基四氢甲烷喋呤。此外,氢营养型产甲烷菌参与了完整的CO2还原途径的所有通路(图3-B)。涉及这些通路的重要酶包括:四氢甲烷蝶呤甲酰转移酶(hfr)、次甲基甲酰四氢甲烷蝶呤环化水解酶(mch)、亚甲基四氢甲烷蝶呤脱氢酶(mtd)以及5,10-亚甲基四氢甲烷蝶呤还原酶(mer)等[63]。此后氢营养型产甲烷途径与乙酸营养型产甲烷途径相同,经四氢甲烷蝶呤S-甲基转移酶(mtr)和甲基辅酶M还原酶(mcr)的催化下形成甲烷[40]。另外,一些新的生化途径被发现,包括DIET[25],膜结合的反向电子转移,以及以氢和/或甲酸盐的形式处理电子的胞质凝聚型酶[64]。厌氧消化的代谢过程的“面纱”正逐渐地被揭开。

图3 产甲烷途径
4.3 功能生物标记物鉴定
随着宏基因组测序技术的广泛使用,越来越多的厌氧消化相关的功能生物标记物被鉴定出来,从而简化了基因组研究。mcrA基因的a亚基是研究最多的产甲烷菌的功能基因,其拷贝数[65]和转录本[66]与甲烷产量存在显著正相关,被认为是厌氧消化中最重要的生物标志物。辅酶F430作为mcrA基因的辅酶同样可以量化产甲烷潜力[66]。Fe-Fe-氢化酶大亚基的基因(hydA)被用作产氢发酵细菌生物标记物[67]。同型产乙酸细菌以甲酰四氢叶酸合成酶编码基因(FTHFS)(Wood-Ljungdahl通路的关键酶)作为生物标志物[68]。此外,基因组研究还可鉴定编码关键微生物基因组的特定代谢途径的酶,包括碳水化合物利用、脂肪酸降解、氨基酸发酵等[69]。通过16S rRNA基因测序与已培养的相似物种做比对,也可以推测出厌氧消化过程的功能[7]。但由于密切相关的生物可能在功能上有所差异,因此应谨慎做出结论[70]。如果基于相关性分析来建立假设,并且使用补充技术进一步论证,得出的结果将更加可靠。
4.4 抗生素耐药基因分析
研究沼渣中抗生素耐药基因(Antibiotic Resistance Genes,ARGs),可以界定其给土壤等其他环境因素可能带来的风险[71]。沼渣中的ARGs可能会传播给致病菌,从而降低致病菌在医疗过程中对抗生素的敏感性,对公众健康构成威胁。ARGs可通过水平基因转移在不同微生物种群间传播,形成具有抗生素耐受性的细菌。使用宏基因组测序不仅揭示了微生物水平基因转移的证据,也证明了其耐药性传播机制。Lee等[72]对具有代表性的有机废弃物(粪便、污泥和食品垃圾回收废水)中ARGs的多样性和数量进行研究,发现粪便中ARGs的总量最高,其次是污泥和食品垃圾回收废水。不同基质间ARGs的多样性和作用机制存在较大差异。研究发现,厌氧消化能有效地降低ARGs拷贝数,但不同抗性机制下的相对丰度则存在明显差异[72]。某些细菌(如Clostridium和Nitrosomonas)可能是多个ARGs的潜在宿主。Luo等[39]发现,在以粪肥和工业废料为原料的反应器中,ARGs的总丰度在7×10-3-1.1×10-1拷贝数/ 16S rRNA基因拷贝数之间变化,并且嗜热型厌氧消化器在去除ARGs上有一定优势。除此之外,延长固体停留时间以及添加生物炭都可以降低ARGs的环境风险[73]。
5 总结与展望
厌氧消化是一种生态友好的有机废弃物处理及资源回收过程。在过去的十年中,随着微生物组学的快速发展,我们对厌氧消化中微生物菌群的结构和功能的认识也有了质的提升。这不仅能够更好地建立微生物群落模型,而且也将使人们更清楚地了解相关的环境条件、代谢途径和演替规律,从而解释这种格局。微生物组学研究为曾经被认为是“黑盒”的复杂微生物群落提供了光明。本文综述了目前微生物组学的类型和常用的生物信息学分析手段,以及最近的研究进展,有助于确定最佳的沼气生产因素的组成,改进厌氧消化器的操作参数;也有助于充分利用厌氧微生物群落潜力,为设计最佳厌氧消化工艺提供理论依据。
目前为止,通过厌氧消化微生物组来改善厌氧消化产甲烷效率仍面临许多挑战。首先,微生物组的厌氧微生物群落研究仍处于起步阶段,需要储存已有数据作为参考。并且新的研究需要读入大量的微生物组数据,为之前的研究结果提供统计支持,这导致需要为测序数据提供庞大的存储空间;其次,大多数序列分析需要与参考数据库中相似序列进行比对,但仍有部分基因在现有数据库中无法匹配到相似序列。未来有必要扩大参考数据库,建立能够对基因组学数据进行全面和综合分析的平台;最后,未来的研究不仅需要考虑微生物的代谢,还需要考虑微生物代谢物如何改变自身或其他微生物的基因潜力与表达,整合多组学分析也是必不可少的。总而言之,要想通过厌氧微生物群落的微生物组学信息来精确调节厌氧消化反应器的性能,仍有很长的路要走。而研究微生物群落如何响应性能参数,已经是向目标迈出了第一步。
