组蛋白去乙酰化酶抑制剂(HDACi)对小麦基因编辑效率的影响及转录组学分析
2021-01-22代文双刘会云杜庆国邹枨王轲
代文双 刘会云 杜庆国 邹枨 王轲
(中国农业科学院作物科学研究所,北京 100081)
作为重要的粮食作物,小麦在世界各地被广泛种植。我国作为世界上最大的小麦生产国和消费国,其生产对保障粮食安全有重要的现实和战略意义。小麦的增产和品质改良愈发重要,产量和品质等重要性状都是受多基因和环境互作的复杂性状,单纯靠常规育种技术很难满足需求,基因编辑技术是改善这一现状的有效手段。基因组编辑技术的发展和应用为植物功能基因研究和作物遗传改良提供了重要的技术支持。与其他基因组编辑技术相比,近年来诞生的CRISPR/Cas(Clustered regularly interspaced short palindromic repeats/Cas)系统(主要包括CRISPR/Cas9和CRISPR/Cas12a)具有操作简单、效率高的优点。自2013年CRISPR/Cas9首次应用于真核生物[1-2],即成为生命科学领域的革命性工具。CRISPR/Cas9系统已成功地用于各种真核生物的基因组工程中,并为基因治疗和植物育种提供了强大的工具[3]。目前,CRISPR/Cas9在植物基因组编辑中的应用主要包括基因功能研究和作物遗传改良。
CRISPR/Cas9系统编辑效率在不同物种间差异很大,而且对于单倍体和多倍体植物来说该技术的难度也存在显著差异[4-5]。比如二倍体植物水稻CRISPR/Cas9技术产生突变的效率可达到80%以上[6]。多倍体植物尤其是小麦、马铃薯等,构建有效的基因编辑系统仍然是当前研究的难点[7-8]。小麦是由A,B和D三个基因组组成的六倍体,基因组约17 Gb,约为水稻的40倍[9-12],且重复序列高达85%-90%[13-14]。利用CRISPR/Cas9技术同时编辑小麦基因组中的多个拷贝依然非常困难,这是限制小麦基因编辑应用的主要原因。虽然在小麦中TaWaxy基因编辑效率高达80%,但3个同源基因同时编辑的效率仅有30%左右[15]。因此,提高小麦尤其是3个基因组的编辑效率的研究工作至关重要。
真核生物染色质结构会影响CRISPR/Cas9基因编辑效率。在真核细胞中细胞核DNA以约147 bp的DNA绕八聚结构的组蛋白1.67圈,进而形成核小体[16-17]。相邻核小体的连接区由10-80 bp的游离DNA与组蛋白H1共同构成,此串联结构进一步折叠、凝聚形成染色质[18]。核小体(染色质的基本结构单位)的存在会阻碍Cas9蛋白对其缠绕DNA上的靶点的连接与切割,降低编辑效率[19-21]。染色质主要以开放和非开放两种状态存在。核小体与DNA缠绕致密、染色质紧实度高的区域为非开放区,反之即为染色质开放区。开放区域染色质状态相对松散,该区域内的基因组占总DNA序列的2%-3%,且超过90%的开放区域与转录因子的结合相关[22]。Wu等对哺乳动物Cas9的结合位点进行了全基因组作图,发现Cas9蛋白与靶基因的作用主要集中在染色质开放区域[23-24]。Chari等[25]在对人类细胞研究中也发现CRISPR/Cas9诱导的插入与缺失在染色质开放区域更多。除在动物中,在植物的研究中也发现这一现象,结合已有的基因组数据信息在水稻开放区域的CRISPR/Cas9编辑效率要高于非开放区域[26]。
研究人员可以通过改变染色质局部结构调控染色质的开放与非开放状态来提高CRISPR/Cas9对基因组的编辑效率。Isaac等[27]利用两种染色质重塑 因 子SNF 2h(Sucrose non-fementing 2h)和RSC(Remodels the structure of chromatin)增加开放染色质区域,进而显著提高了Cas9蛋白靶向DNA能力。HMGN(High mobility group proteins)、HMGB1(High mobility group box 1 protein)组蛋白H1和染色质重塑复合物等是能与蛋白质自然构象相互作用的蛋白质。其中HMGN蛋白是核小体连接蛋白,可以调节染色质结构[28-30];HMGB1具有DNA弯曲活性,并且与组蛋白H1竞争核小体附近的染色质[31-33]。一种名为CRISPR-chrom的方法[34],通过将Cas9直系同源物与这些染色质调节相关的多肽融合在一起,能显著提高Cas9的编辑效率。简单来说,这些方法都是通过重塑染色质,即调整或移除核小体、调节组蛋白等来提高Cas9对靶点的编辑效率。
组蛋白乙酰化在核小体重塑中起主要作用[35-36]。它是研究最早,特征最多的翻译后修饰[37]。组蛋白乙酰化修饰可以改变染色质结构,调控基因转录、DNA复制和修复[38-39]。组蛋白乙酰化的赖氨酸可被重塑因子SWI2/SNF2(Switching defective2/sucrose non-fermenting2)识别和结合进行染色质重塑[40]。其修饰水平受组蛋白乙酰化转移酶(Histone acetyltransferases,HAT)和组蛋白去乙酰化转移酶(Histone deacetylases,HDAC)的调节,其中组蛋白去乙酰化转移酶在植物的生长、发育和响应胁迫方面发挥重要作用[41-44]。
丁酸钠(Sodium butyrate)和尼克酰胺(Nicotinamide)是常见的用来研究组蛋白乙酰化修饰水平的两种组蛋白去乙酰化酶抑制剂(Histone deacetylase inhibitors,HDACi)。丁酸钠是一种短链脂肪酸,可抑制RPD3/HDA1 HDAC基因家族的同源物[45]。尼克酰胺是维生素B3的衍生物,属于SIR2类的 HDAC去乙酰化酶抑制剂,当过量加入会显著抑制哺乳动物和酵母中HDAC[46-47]。尼克酰胺处理会抑制拟南芥产生SIR2类的HDAC(SRT1和SRT2),导致染色质重塑蛋白VIN3(Vernalization Insensitive 3)位点的表达量升高[48-49],进而抑制FLC(Flowering Locus C)的表达。FLC水平的升高会抑制开花,即尼克酰胺处理对FLC的抑制作用可促进开花[49-51]。在玉米中也有HDACi的应用,Tiricz等[52]用丁酸钠和尼克酰胺处理玉米细胞,可以促进染色质区域松散状态,而且能够提高寡核苷酸 定 向 诱 变(Oligonucleotide-directed mutagenesis,ODM)的效率,即提高ODM类基因编辑效率。
本研究主要利用组蛋白去乙酰化酶抑制剂处理小麦幼苗,通过对小麦幼苗的生长及转录组学分析,发现了6个甲基转移酶合成通路基因下调表达都与染色体开放状态相关,从而发现尼克酰胺处理会改变小麦染色质状态及基因表达;我们进一步以对未发生编辑的TaAGO4a基因编辑转基因小麦材料的T1代为材料,取其开花14 d的幼胚进行一步成苗并利用尼克酰胺处理T2代幼苗,结果显示尼克酰胺确实可以增加小麦TaAGO4a基因编辑效率。这为提高小麦基因编辑效率提供一种新策略,为进一步研究小麦CRISPR/Cas9系统提供科学依据。
1 材料与方法
1.1 材料
转录组测序所用材料为六倍体小麦春性品种Fielder,以含有SpCas9和针对TaAGO4a基因的sgRNA且未发生编辑的Fielder的T1代转基因植株为材料研究尼克酰胺与编辑效率之间关系的实验。由中国农业科学院作物科学研究所转基因中心小麦遗传转化实验室提供。
1.2 方法
1.2.1 植物培养 选取大小一致、饱满的小麦种子生长于温室中,日照16 h,24℃;黑暗8 h,18℃。培养条件为光强300 μmol/m2/s,湿度为45%,每周浇水一次。选取开花授粉后14 d左右未成熟的小麦籽粒,用70%的乙醇洗1 min,然后用15%的次氯酸钠震荡洗涤10 min,最后用无菌水洗3次。于超净工作台中无菌操作分离小麦幼胚,将小麦幼胚的胚轴朝上放置于1/2 MS(2.215 g MS/L 粉末,20 g/L蔗糖,0.5 g/L MES,3 g/L 植物凝胶,pH 5.8)培养基中培养。分别设置浓度为0 mmol/L,2.5 mmol/L,5 mmol/L的尼克酰胺培养基和0 mmol/L,5 mmol/L,10 mmol/L的丁酸钠6种培养基。长日照培养(16 h光照/8 h 黑暗)室温25℃,光强300 μmol/m2/s。
1.2.2 生理指标测定及样品选取 记录尼克酰胺和丁酸钠两种试剂不同浓度处理的小麦幼苗长势,记录生长7 d、14 d时小麦幼苗的株高与生长状况。对3种浓度尼克酰胺处理的小麦幼苗,每种材料取3个重复,蒸馏水冲洗3-4次,吸干表面水分,液氮速冻,置于-80℃暂存。取T1代转基因植株的幼苗叶组织约0.1 g提DNA,确认这批材料是含未被编辑的TaAGO4a基因且Cas9/sgRNA编辑复合体仍然存在的植株。对上述材料继续培养并摘取其幼胚进行尼克酰胺处理,并对处理7 d和14 d的T2代幼苗叶组织取约0.1g提DNA,检测植株编辑情况。
1.2.3 转录组测序 利用TransZolUp(TransGen Biotech,USA)提取不同浓度的尼克酰胺(0 mmol/L,2.5 mmol/L,5 mmol/L)处理7 d和14 d叶组织的总RNA,实验设置两个重复共12个样本,分别记做ctrl7d_1,ctrl7d_2,t7d-2.5_1,t7d-2.5_2,t7d-5_1,t7d-5_2,ctrl14d_1,ctrl14d_2,t14d-2.5_1,t14d-2.5_2,t14d-5_1,t14d-5_2。将这些RNA去除基因组DNA后,检测RNA样本的浓度与完整性,浓度均> 800 ng/μL,RIN > 7(RNA完整性,大于7既满足建库要求,大于6.3可以获得较高的mapping率),OD260/280> 1.8,RNA质量检测合格,即可送北京贝瑞和康生物技术有限公司进行RNA-seq高通量测序。
1.2.4 数据分析 利用Trimmomatic软件[53]对这24个文库Raw Reads去除测序接头、低质量Reads、未知碱基比例大于5%的Reads以及接头污染的Reads,得到Clean Reads。利用FastQC软件对Clean Reads进行质控,通常Q30(碱基测序错误率小于0.1%)的碱基数占总碱基数的85%以上,才认为数据符合分析要求。利用Hisat2 v2.1.0[54-55]将过滤合格的Clean Reads比对到小麦RefSeq v1.0[12]参考基因组上,双端比对的最大片段长度设为400 bp,其他参数选用默认参数,比对结果存储于Sam(Sequence alignment map)文件中。用Samtools软件把Sam转为Bam(Binary alignment map)格式文件,参数采用-sort -@ 8。然后利用Stringtie v1.3.3[55-56]软件组装、拼接转录本,参数使用-B -G。输出文件用R语言中的Ballgown包来计算基因表达量,结果以FPKM(Fragments per kilobase of exon per million fragments mapped,每千碱基外显子百万片段数)表示基因的表达水平。Htseq-count v0.8.0[57]软件用以计数bam文件中的reads数。差异表达分析选用R包DEseq2,输入文件正是上述Htseq-count的输出文件。其中FKPM > 1被定义为“表达基因”,否则认为基因非表达。矫正P值< 0.05及差异变化倍数(|Log2 Fold Change|,LFC)≥ 2被定义为差异表达基因。
1.2.5 GO富集与pathway分析 差异表达基因的GO富集分析利用R语言中的clusterProfiler包完成。小麦RefSeq v1.0参考基因组基因的GO注释以及GO号的注释下载自:EnsemblePlants(http://plants.ensembl.org/index.html),pAdjustMethod:”BH”。获得的主要基因利用Plant reactome(https://plantreactome.gramene.org/index.php?lang=en)网站进行pathway富集分析。
1.2.6 T1代阳性转基因植株鉴定 对T1代未发生编辑的QA147-1,QA167-2和QA167-7三个株系的TaAGO4a基因编辑材料幼苗进行不同浓度的尼克酰胺处理,在处理7 d时取样,然后处理14 d时再次进行取样,对取得的样品用DNA提取试剂盒(康为世纪生物科技有限公司)从植株叶片中提取基因组DNA,并用barF(ACCATCGTCAACCACTACATCG)和barR(GCTGCCAGAAACCACGTCATG)引 物 对bar基因进行PCR检测,反应程序为95℃预变性10 min;95℃变性30 s,64℃退火30 s,72℃延伸30 s,扩增25个循环;72℃终延伸5 min。PCR扩增产物在1%琼脂糖凝胶上进行电泳分离,筛选阳性转基因植株。
1.2.7 T1代和T2代转基因植株突变类型检测 首先利用TaAGO4a基因的通用引物AGO4a486F和AGO4a1404R对阳性的TaAGO4a基因编辑转基因材料进行聚合酶链式反应-限制性内切酶图谱分析(PCR-RE)检测突变类型。PCR-RE具体步骤是首先利用普通2×Taq MasterMix,扩增阳性转基因植株的靶点序列(GCACACGCAAGAAGCCATTC),然后对扩增产物用相应的限制性内切酶XmnI酶切2 h,最后用浓度为2%的琼脂糖凝胶进行电泳分离,根据酶切片段的大小和数目判断转基因植株的编辑类型。然后对于编辑植株进一步利用3A,3B和3D三个基因组的特异性引物进行PCR-PE实验,其中3A基因组特异性引物为AGO4a486F和Ago4aA1629R;3B基因组为Ago4aB749F和AGO4a1404R;3D基因组为Ago4aD698F和AGO4a1404R,并对酶切后的PCR产物送由北京擎科生物科技有限公司进行突变体测序,所用引物序列见表1。

表1 PCR引物序列表
2 结果
2.1 组蛋白去乙酰化酶抑制剂(HDACi)对小麦幼苗生长的影响
丁酸钠处理的小麦幼苗在7 d和14 d时几乎都不能生长(图1-A),丁酸钠严重抑制小麦的生长,因此不适合用于小麦幼苗的处理。尼克酰胺处理在初期(7 d内),小麦幼苗生长落后于野生型对照组,但差异不显著。而且2.5 mmol/L和5 mmol/L两个浓度处理之间的差异也不显著(图1-B和1-D)。随着处理时间的延长,处理组与对照组在14 d时无显著差异(图1-C和1-D)。因此尼克酰胺处理对小麦生长的影响较小。
2.2 测序数据质控与比对情况评估
RNA-seq的建库质量及测序质量会对后续实验分析产生较大影响,因此对测序数据的质控与评估十分重要。尼克酰胺(0 mmol/L,2.5 mmol/L,5 mmol/L)处理7 d和14 d每个处理设置两个重复共12组样本,共95 Gb下机数据。利用Trimmomatic数据过滤共获得约5000万个Clean reads;FastQC软质控结果显示Q30大于92%;将Clean reads比对到小麦RefSeq v1.0参考基因组,样本完全比率均在90%以上(表2)。质控及比对结果显示下机数据可以很好的满足后续分析的需求。

图1 不同的组蛋白乙酰化抑制剂对小麦幼苗生长的影响

表2 测序数据质控与比对情况统计
2.3 相关性分析
利用R语言绘制PCA图展示各样本间以及同一处理不同重复的相关性(图2)。第一个主成分PC1占投影特征的方差比例为60%,第二个主成分PC2占了23%的方差比例,两者一起可以满足我们的阈值。而且可以直观的看到同一处理不同重复聚类在一起,说明重复间相关性较高,相同的时间处理也存在相关性。

图2 主成分分析图
2.4 基因差异表达分析
利用R语言的DEseq2包研究不同尼克酰胺处理的差异表达基因,依据校正后的P-value < 0.05和差异变化倍数≥ 2,即2倍的差异变化的规则来定义差异表达基因。其中LFC > 0表示基因表达上调,LFC < 0表示基因表达下调。结果显示,t7d-2.5和t7d-5分别与ctrl7d相比时差异表达基因有997和863个,其中有791和662个上调基因;另外,t14d-2.5和t14d-5与ctrl14d相比有144和318个差异表达基因,其中上调基因的数目分别是88和148个(图3-A),t7d-2.5组中差异表达基因数目最多,t14d-2.5组最少。相同浓度下(2.5 mmol/L或5 mmol/L),尼克酰胺处理7 d的差异表达基因数目远多于处理14 d,说明处理前期对小麦幼苗影响更大,这个结果与尼克酰胺处理14 d时小麦幼苗的生长状态基本恢复到对照组一样完全一致,因此与尼克酰胺处理相关的基因应该可以限定在处理7 d的差异基因中。处理时间相同情况下,t7d-2.5与t7d-5差异表达基因在数目上相差较少,且两组有700个基因是相同的;而处理14 d,t14d-5组中差异基因数目比t14d-2.5多一倍,其中113个基因是相同的(图3-B)。

图3 差异表达基因分析
2.5 GO富集与pathway富集分析
GO(Gene ontology)即基因功能富集分析,是对基因在不同维度和不同层次上的描述。对基因的描述一般从3个层面进行:Cellular component(CC,细胞成分)、Biological process(BP,生物学过程)、Molecular function(MF,分子生物功能)。为了进一步研究不同处理时间对小麦转录组的影响,我们对7 d(t7d-2.5和t7d-5)和14 d(t14d-2.5和t14d-5)的所有差异表达基因进行GO富集分析(图4,表3)。
对GO富集分析图中主要terms进一步统计(表3)。尼克酰胺直接相关的“nicotianamine synthase activity term”、“nicotianamine biosynthetic process term” 集中出现在幼苗处理7 d,处理14 d未富集到这两条terms,且上述两terms内基因均下调表达,说明处理7 d以内的幼苗对尼克酰胺更加敏感。相比于7 d,14 d富集到的GO term主要为小麦生长常见term。尼克酰胺处理7 d的细胞分裂“cell division term”基因相对于对照组均表达上调,而处理7 d的甲基转移酶“O-methyltransferase activity term”的基因则表现为下调表达。“transaminase activity term”氨基转移通路基因也在7 d处理后相对于对照组表达上调。据统计,上述主要terms中所含基因共25个(不含重复)。
我们对上述中的terms中所涉及的基因使用Plant Reactome数据库进行pathway富集分析。通过整合基因组、生化系统和化学分子等方面的数据,系统的分析基因产物功能,根据功能将这些注释的pathway分为细胞过程、遗传信息处理、环境信息通路、新陈代谢和生物有机系统5个类别(图5)。结果发现“transaminase activity term”内基因关联到多条与氨基酸代谢相关的pathway,其中赖氨酸合成VI通路包含在内(图5)。
2.6 T1代TaAGO4a基因编辑材料阳性检测及突变类型检测
对T1代 分 别 来 自QA147-1,QA167-2和QA167-7三个株系的TaAGO4a基因编辑材料幼苗取样提DNA,并进行PCR和PCR-RE检测。共检测47株材料,PCR扩增bar基因发现39株转基因植株为阳性(图6-A)。利用通用引物对AGO486F和AGO1404R扩增这些阳性转基因植株的靶点序列并用限制性内切酶XmnI酶切对PCR产物酶切(PCRRE),检测发现,39株植株TaAGO4a基因均未发生编辑(图6-B)。

图4 差异表达基因(DEGs)GO富集分析

表3 主要GO trems统计表
2.7 尼克酰胺处理后T2代TaAGO4a基因编辑材料的阳性检测和突变类型检测
我们对上述T1代TaAGO4a基因编辑材料的阳性植株的幼胚接种到培养基上进行尼克酰胺处理实验,总共接种了69个幼胚并全部成苗,0 mmol/L,2.5 mmol/L和5 mmol/L培养基上分别有19、32、18株苗。上述材料培养至7和14 d时分别进行两次取样检测,根据尼克酰胺处理时间与浓度共分为6组样品(7-CK,7-2.5,7-5,14-CK,14-2.5,14-5)。首先对这些T2代植株检测,发现42株为转基因阳性植株(图7-A),0 mmol/L,2.5 mmol/L和5 mmol/L培养基上分别有12、18和12株阳性。利用TaAGO4a基因通用引物(AGO4a486F和AGO4a1404R)对阳性植株进行PCR-RE检测,结果显示0 mmol/L、2.5 mmol/L和5 mmol/L处理7 d时都没有检测到编辑植株,0 mmol/L和2.5 mmol/L处理14 d时也没有检测到编辑植株,只有5 mmol/L浓度处理14 d检测到1株杂合编辑植株(图7-B),编辑效率为8.3%(表4)。

图5 主要基因的Pathway分布图
为了进一步检测这株突变体材料的的编辑类型,我们分别利用3A,3B和3D三对特异性引物扩增靶点序列(图8),PCR-RE结果显示该材料发生编辑的是3A和3B基因组,3D基因组未发生编辑(图8-A),突变体测序显示3A和3B基因组编辑类型均为靶点处存在16 bp碱基缺失(图8-B)。

图6 T1代转基因植株bar基因及突变类型检测情况

图7 14 d T2代转基因植株bar基因及突变类型检测情况

表4 各处理编辑情况统计
3 讨论
3.1 HDACi对小麦幼苗生长的影响
Tiricz等[52]通过检测细胞活力,发现丁酸钠和尼克酰胺处理后的玉米细胞生长活力均未受到阻碍。而Bond等[49]利用丁酸钠、TSA(Trichostatin A)和尼克酰胺3种HDACi处理拟南芥时发现尼克酰胺对拟南芥幼苗生长的影响较小,丁酸钠和TSA相对会抑制拟南芥生长。本实验结果也发现不同的组蛋白去乙酰化抑制剂对小麦幼苗生长的影响不同。不同浓度的丁酸钠处理都严重抑了小麦幼苗的生长。这说明小麦幼苗对丁酸钠反应敏感并不适合用来处理小麦。不同浓度的尼克酰胺处理小麦幼苗,前期(约7 d)幼苗生长较对照组缓慢,但处理至14 d,处理与对照生长并无明显差异。虽然尼克酰胺处理前期会相对影响小麦苗的生长,但随生长发育及小麦幼苗自身的调控作用适应了尼克酰胺处理,即尼克酰胺处理不会阻碍幼苗后续生长。因此用尼克酰胺处理小麦更合适。
3.2 尼克酰胺改变染色体状态候选基因的筛选
组蛋白乙酰化的赖氨酸是SWI2/SNF2亚家族染色质重塑因子结构域识别和结合的位点,组蛋白H3上甲基化的赖氨酸是ISWI亚家族染色质结构域结合和识别的位点[58],即赖氨酸与染色质重塑因子作用相关。组蛋白尾部赖氨酸残基的α-氨基乙酰化是一种动态的染色质修饰。我们通过对主要GO富集转氨基活性terms中差异表达基因的pathway富集分析发现这些参与转氨基作用的基因与赖氨酸合成(Lysine biosynthesis VI)通路关联密切,这说明尼克酰胺处理可能与染色质状态有关。组蛋白修饰相关因子与甲基转移酶(Dnmt)相互作用是组蛋白修饰调控基因表达的重要手段。研究表明Dnmt可与组蛋白去乙酰化酶(HDAC)、共抑制因子(Co-repressor)和ATP依赖的染色质重塑蛋白等结合形成抑制复合物,与组蛋白乙酰化和染色质重塑共同作用,从而抑制基因表达[59-61]。抑制Dnmt的基因表达与组蛋白修饰和染色质重塑相关[62],比如Dnmt3a可与HP1(Heterochromatin protein 1)相互作用,而HP1具有识别甲基化的组蛋白、稳定异染色质浓缩结构的作用。因此,抑制Dnmt3a表达有利于促进染色质开放[63]。本研究的GO富集分析发现,尼克酰胺处理导致6个Dnmt基因下调表达,而Dnmt基因下调有利于染色质开放,因此尼克酰胺处理可能促进染色质开放。开放区域染色质状态相对松散,超过90%的开放区域与转录因子的结合相关且该区域细胞分裂活跃[22]。细胞分裂增强说明DNA复制活动增强,一般来说染色质的开放性在复制后会得到不同程度的恢复[64]。本研究中处理7 d有4个细胞分裂基因表达上调,说明尼克酰胺处理有利于染色质开放,进而有利于转录因子的结合以及基因的复制与转录等表达活动。综上所述,尼克酰胺处理对染色质开放具有促进作用。
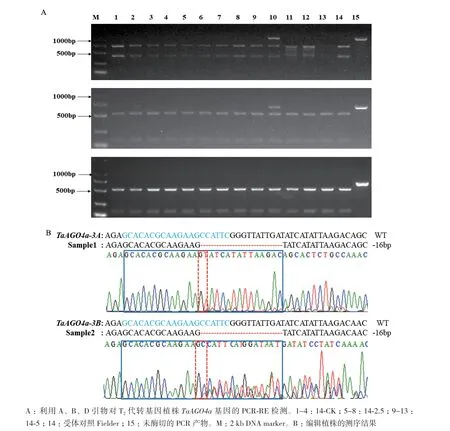
图8 突变体材料编辑类型检测
3.3 尼克酰胺处理和CRISPR/Cas9编辑效率的关系
Tiricz等[52]利用DNase I酶的敏感性发现5 mmol/L尼克酰胺和10 mmol/L丁酸钠处理后染色质状态更加开放,靶基因mGFP与ZmMEK1编辑效率更高。Bond等[49]在拟南芥中发现5 mmol/L尼克酰胺处理14 d诱导VIN3基因表达水平最高。本实验利用尼克酰胺处理TaAGO4a基因的未发生编辑的基因编辑材料,结果显示0 mmol/L,2.5 mmol/L和5 mmol/L处理7 d时都没有检测到编辑植株,而0 mmol/L和2.5 mmol/L处理14 d时也没有检测到编辑植株,只有5 mmol/L浓度处理14 d下检测到1株A和B基因组均杂合编辑的植株,编辑效率从0提高到了8.3%。其中经过对PCR-RE产物测序发现3A基因组缺失了16 bp,而3B基因组则是在靶点处出现套峰,这说明酶切还不够彻底,然后利用DSDecodeM(http://skl.scau.edu.cn/dsdecode/)分析发现3B基因组也缺失了16 bp[65]。因此5 mmol/L处理14 d是尼克酰胺处理小麦的最佳条件,这与之前在玉米和拟南芥中报道的最佳条件基本一致。这是首次在小麦中明确报道尼克酰胺可以促进编辑效率,进一步明确了尼克酰胺提高小麦基因编辑效率的可行性,也为提高小麦编辑效率提供了一种可行的新方法。
4 结论
相较于丁酸钠,尼克酰胺对小麦生长影响较小。尼克酰胺处理有利于小麦染色质趋于开放状态,第一次明确尼克酰胺可以提高小麦基因编辑效率,且5 mmol/L尼克酰胺处理14 d是最佳条件,编辑效率从0提高到了8.3%,而且实现了两个基因组靶点的编辑。
