猪不同发育阶段肠道微生物菌群特征分析
2021-01-20陈宝剑吴永绍覃兆鲜潘天彪关志惠陈少梅吴柱月谢炳坤
陈宝剑,吴永绍,覃兆鲜,张 冰,潘天彪,关志惠,陈少梅,吴柱月,谢炳坤
(广西壮族自治区畜牧研究所,广西家畜遗传改良重点实验室,广西南宁 530005)
猪肠道中微生物主要是厌氧菌和兼性厌氧菌,其中厚壁菌门和拟杆菌门占90% 以上,在维护机体健康、提高机体免疫力、营养物质吸收代谢等方面发挥着重要作用[1]。不同时期猪肠道微生物菌群有显著差异,胚胎时期肠道处于一种无菌状态,分娩过程中受母体产道、粪便以及周围环境的影响开始出现微生物,主要以大肠杆菌、葡萄球菌等为主[2]。哺乳期仔猪肠道中的优势菌群主要为乳酸杆菌和链球菌,仔猪从哺乳期到断奶期间,内源性的乳酸菌、厌氧菌群数量减少,外源性大肠杆菌、沙门菌等致病菌数量增加[3]。仔猪断奶后1~3 周内会形成相对稳定的微生物群落,此时胃肠道微生物优势菌种为双歧杆菌[4]。不同饲料营养成分是引起猪肠道微生物变化的重要原因。Peng 等[5]研究发现,育肥猪日粮蛋白质水平降低5%能够提高结肠微生物多样性;Zhou 等[6]研究发现,生长猪日粮蛋白质水平降低3%后在属水平上多种微生物菌群相对丰度差异显著;Jaworski 等[7]研究发现,饲喂高纤维水平饲粮的猪胃肠道微生物活性明显高于饲喂低纤维水平饲粮。仔猪断奶2~3 周后肠道微生物结构基本形成,此后生长发育和日粮改变是引起猪肠道微生物结构持续变化的主要原因,现代生猪养殖从断奶到出栏会根据不同生长阶段来提供营养需求,必将引起猪胃肠道微生物结构发生改变。虽然关于不同阶段猪肠道微生物菌群的研究已有很多,但有关猪断奶后不同生长阶段与饲料营养水平改变对猪肠道微生物菌群的影响研究尚未见报道。本研究旨在分析断奶后不同生长阶段和日粮营养水平的变化对肠道微生物菌群的影响,更好的揭示饲粮营养调控在猪不同生长阶段对肠道微生物的组成与平衡作用机制。
1 材料与方法
1.1 实验材料 试验猪由广西壮族自治区畜牧研究所种猪场提供,将50 头28 日龄体重8 kg 左右的杜× 长×大断奶仔猪按照体重和性别随机分为5 栏,根据不同的生长阶段选择广西扬翔股份有限公司生产的饲料进行饲喂,其营养成分见表1。分别在第60、90、120、150、180 日龄上午饲喂前,从每栏随机挑选5 头猪,每头采取100 g 左右的新鲜粪样,-80℃保存。
1.2 血清生化指标检测 分别在采集粪便的同时,前腔静脉采血5 mL,3 000 r/min,4℃,离心10 min,吸取血清-80℃保存。利用ELISA 技术测定免疫球蛋白M(IgM)、免疫球蛋白G(IgG)、免疫球蛋白A(IgA)、白细胞介素2(IL-2)、白细胞介素6(IL-6)含量,ELISA 试剂盒均购自上海江莱生物科技有限公司。
1.3 肠道微生物测序 提取粪便总DNA,选择16S rRNA 基因的V4 区作为扩增和测序的目的区间,扩增长度为468 bp,扩增引物序列:338F:5'-ACTCCTAC GGGAGGCAGCAG-3',806R:5'-GGACTACHVGGGT WTCTAAT-3',将扩增后的片段回收,利用PE300 测序平台进行测序。
1.4 肠道微生物菌群特征分析
1.4.1 测序数据优化 MiSeq 测序得到的是双端序列数据,首先根据PE reads 之间的相互重叠关系,将成对的reads 拼接成一条序列,同时对reads 的质量和拼接的效果进行质控过滤,根据序列首尾两端的条形码和引物序列区分样品,得到有效序列,并校正序列方向。
1.4.2 OUT 聚类分析 利用Usearch 平台vsesion 7.0软件进行OUT 聚类分析,首先对优化后的序列提取非重复序列,并去除没有重复的单序列,按照97% 相似性对非重复序列进行OTU 聚类,在聚类过程中去除嵌合体,得到OTU 的代表序列,将所有优化序列对比至OTU 代表序列,选出与代表序列相似性在97%以上的序列,生成OUT 数据。
1.4.3 肠道微生物Beta 多样性分析 根据beta 多样性距离矩阵进行层级聚类分析,使用UPGMA 算法构建树状结构,利用Qiime 软件计算beta 多样性距离矩阵,然后用R 语言做出树状图,Vegan 软件包制作NMDS 分析图。
1.4.4 肠道微生物多样性指数分析 分别计算每个单独样本的Chao、PD、Ace、Sob 指数和Coverage 多样性指数,通过每个样本的多样性指数分析来反映出整个微生物群落的丰富度、多样性和覆盖率,然后对不同组间多样性指数进行t检验。
1.4.5 肠道微生物物种间差异分析 根据得到的群落丰度数据,运用克氏秩和检验,对不同组微生物群落之间的物种进行假设检验,评估不同组间物种丰度差异的显著性水平。
1.4.6 肠道微生物不同分类学水平上的群落组成 利用现有细菌和古菌16S rRNA silva 数据库(Silva(Release128 http://www.arb-silva.de),Greengene(Release 13.5 http://greengenes.secondgenome.com/);真菌18S rRNA 数据库:Silva(Release128 http://www.arb-silva.de);真菌ITS数据库:Unite(Release 7.0 http://unite.ut.ee/index.php)对每个OTU 所对应的物种进行分类,采用RDP classifier贝叶斯算法对97%相似水平的OTU 代表序进行分类学分析,并分别在域、门、纲、目、科、属、种水平上统计每个样品的群落组成。
1.4.7 肠道微生物多样性与环境因子关联分析 使用R软件vegan 包对血清生化指标与肠道差异菌属关联分析。通过计算环境因子与所选菌属之间的Sperman 相关系数,将获得的数值矩阵通过Heatmap 图直观展示。
1.5 统计分析 试验数据先经Excel 统计,利用SPSS 18.0 软件对不同日龄间血液生化指标进行单因素方差分析,采用 Duncan's 法进行多重比较检验,结果用平均值±标准差表示。
2 结果与分析
2.1 猪不同发育阶段血清生化指标检测 由表2 可见,随着仔猪的生长发育,血清生化指标出现显著变化,特别是在饲喂的前2 个月内,血液中IL-2、IL-6、IgG、IgM、IgA 浓度与其他阶段都差异显著,此时猪肠道微生物还处于一个不稳定的状态。IgG 和IgA 在不同时期差异显著。仔猪在生长过程中,其体内免疫能力也随着饲料和环境的变化而改变。
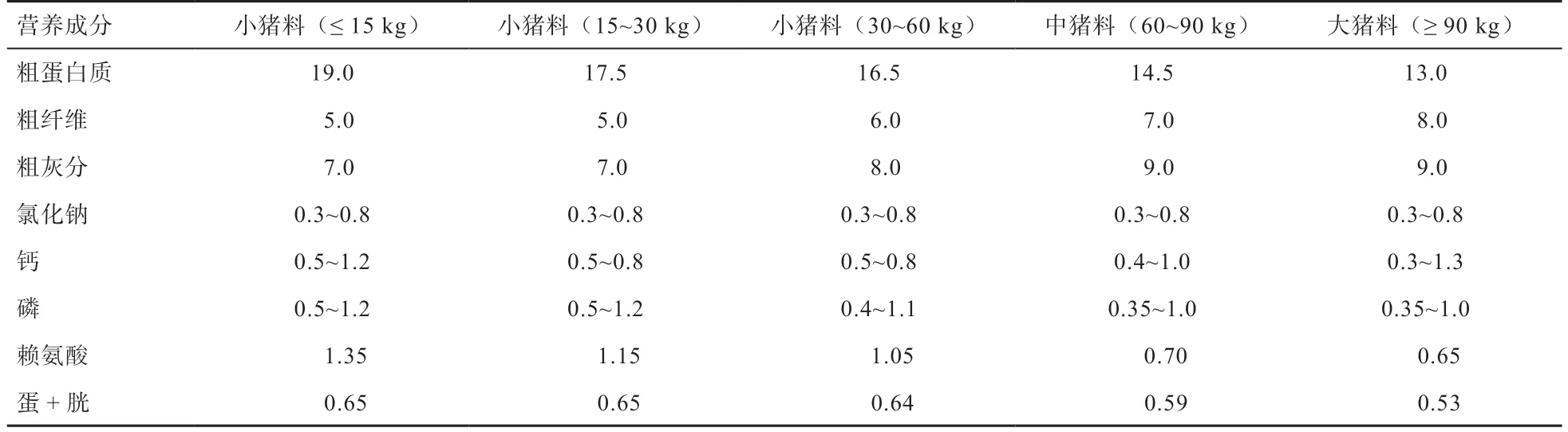
表1 日粮营养成分 %
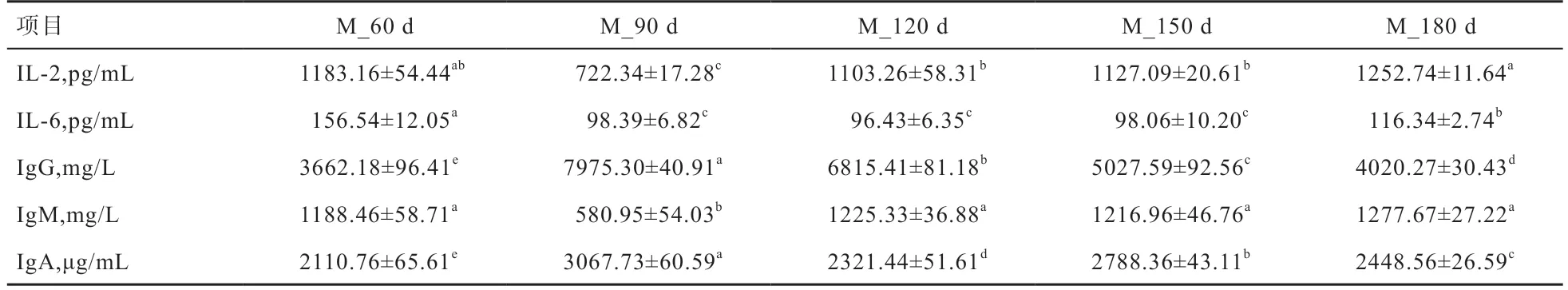
表2 血清生化指标检测结果
2.2 肠道微生物DNA 的提取及数据筛选 25 个样本均获得了质量良好的基因组DNA,同时也扩增出了符合目的片段大小的PCR 产物,测序完成后,对所测数据进行质控检查。由图1 可以看出,样本的Shannon 和Sobs 稀释曲线,当样品随机抽取的数据量为35 000 左右时,曲线开始趋于平坦,表明取样深度基本一致,稀释曲线均达到平台期,包含了样本中绝大多数微生物多样性的信息。数据经优化整理后,每个处理组的数据统计结果(表3)。各组间所获得的序列条数及碱基个数经统计学检验均差异不显著,5 个处理组序列长度均集中在421~440 bp(51.64%)和441~460 bp(占48.36%),每个处理组的平均长度同为440 bp。5 个试验组共获得1 612 个OUT,其中60 日龄时获得了947 个、90 日龄时获得1 169 个、120 日龄时获得1 133 个、150日龄时获得1 261 个、180 日龄时获得1 217 个。由组物种Venn 图(图2)所示,5 个试验组共享了607 个OTU,有4 个试验组共享了338 个OUT,3 个试验组共享了230 个OUT,2 个试验组共享了213 个OUT,60 日龄组有85 个专有OTU,90 日龄组有50 个专有OTU,120 日龄组有16 个专有OUT,150 日龄组有33个专有OTU,180 日龄组有36 个专有OTU。
2.3 肠道微生物NMDS 分析和聚类分析 对5 个试验组共计25 个样本进行NMDS 及聚类分析,每个试验组5个体间的微生物群落组成相似(图3-A),同一组个体均聚在同一个大的分支上(图3-B),表明本试验中所采集的样品重复性较好,组间菌群差异明显。
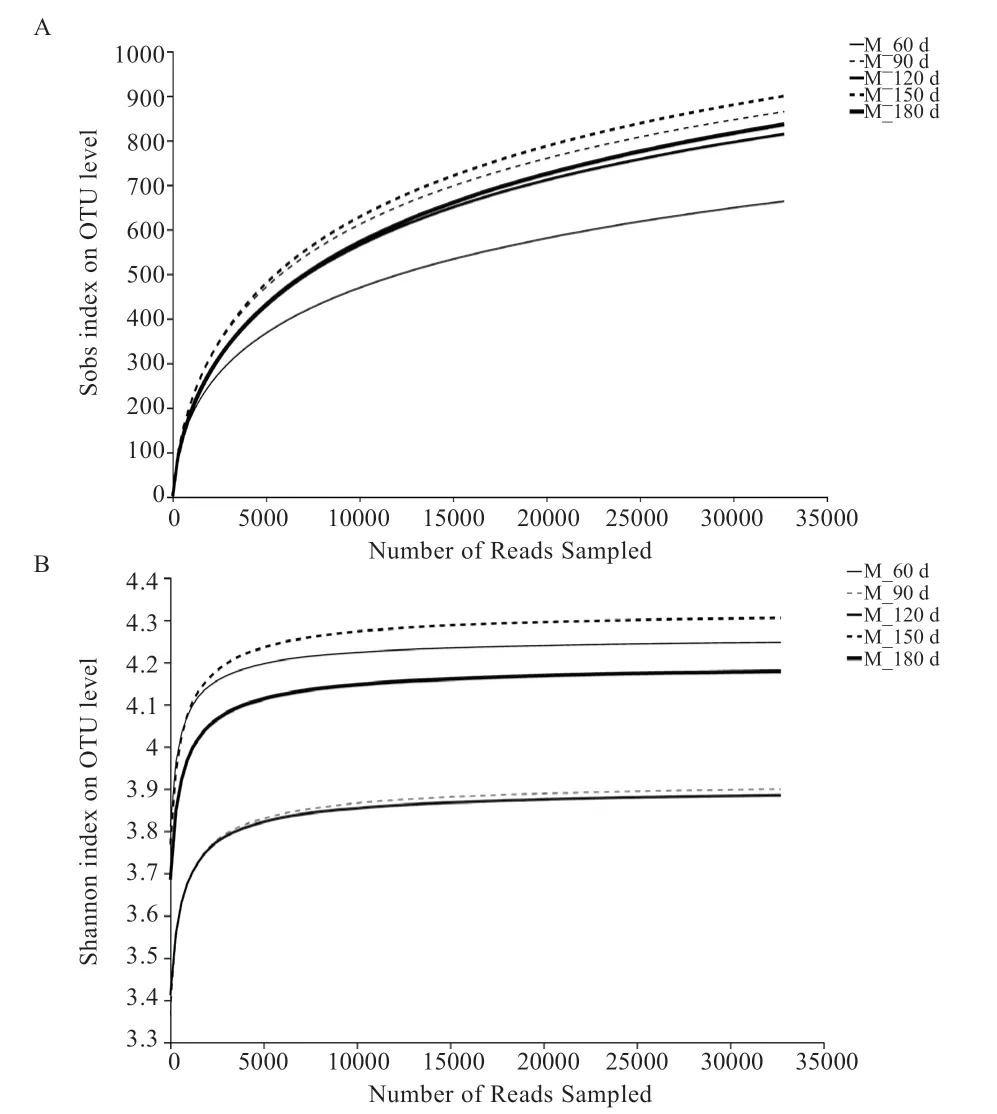
图1 Shannon(A)和Sobs(B)稀释曲线分析结果
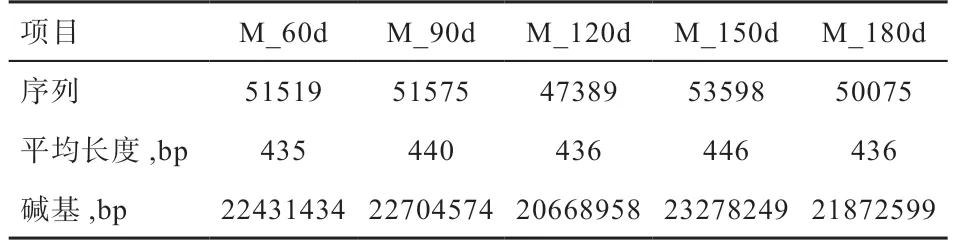
表3 样本测序结果统计表
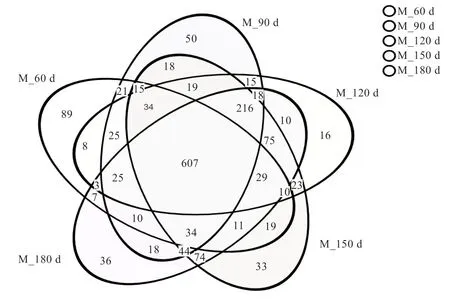
图2 不同试验组物种Venn 图
2.4 猪肠道微生物多样性分析 通过对每个样本的Chao、PD、Ace、Sobs 等多样性指数的计算,并对不同组间多样性指数进行t 检验分析(图4)后发现,Chao、Ace、Sobs 指数的变化趋势基本一致,在60 日龄与其他时间均差异极显著,说明随着仔猪的生长,猪肠道微生物多样性显著升高,达120 日龄后,猪肠道微生物趋于稳定,组间差异变小。全部25 个样本的覆盖率均在99%以上,Coverage 指数随着仔猪体重的增加而减少,说明随着仔猪的生长,猪肠道微生物的种类数有所减少。
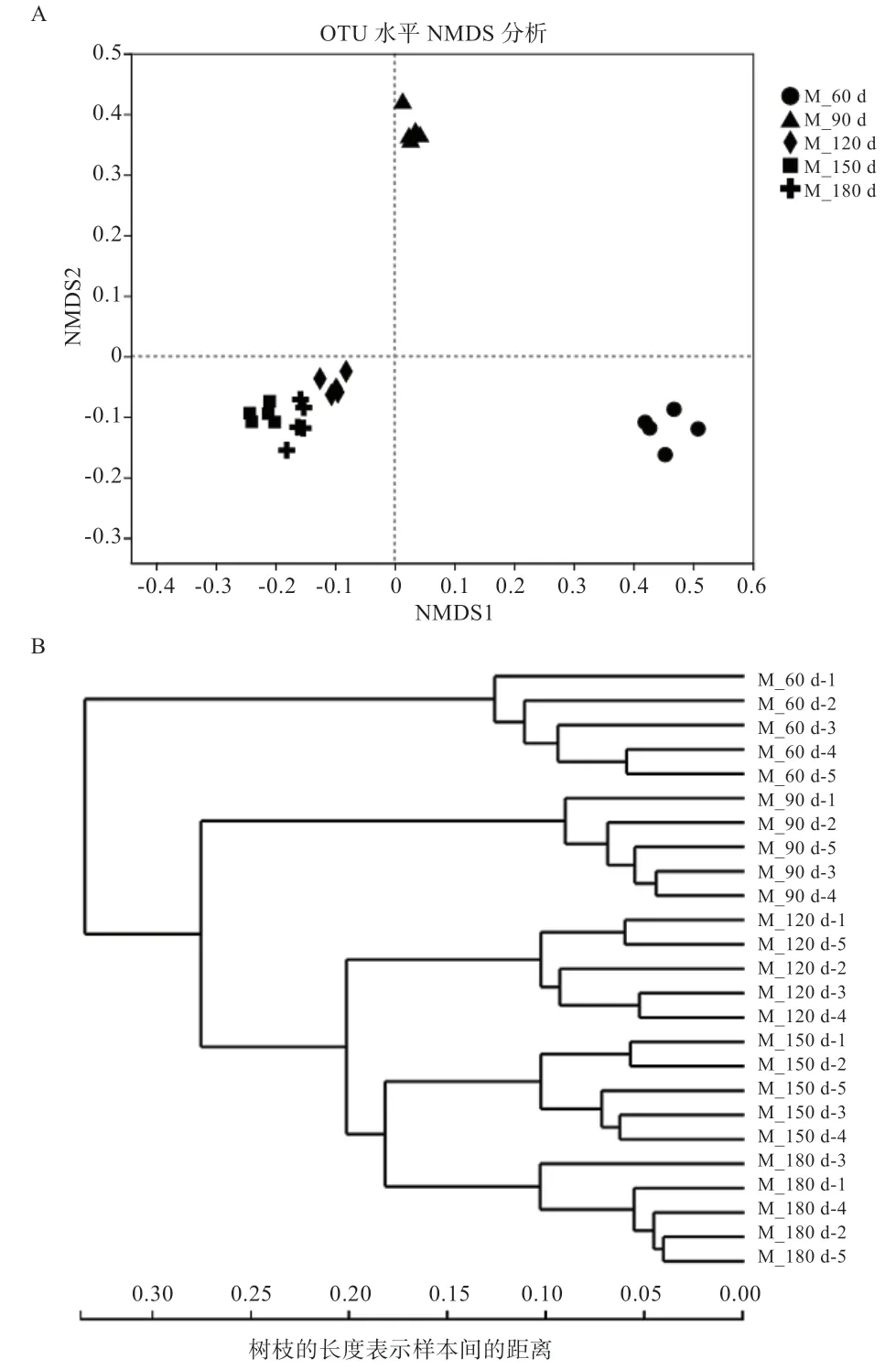
图3 不同样品NMDS 分析图及相似度树状图
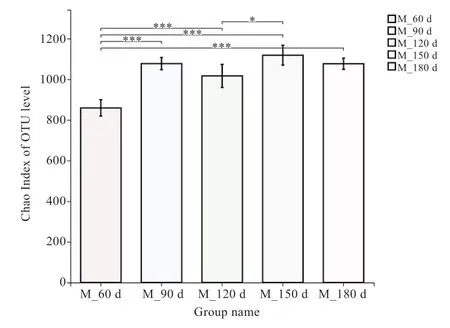
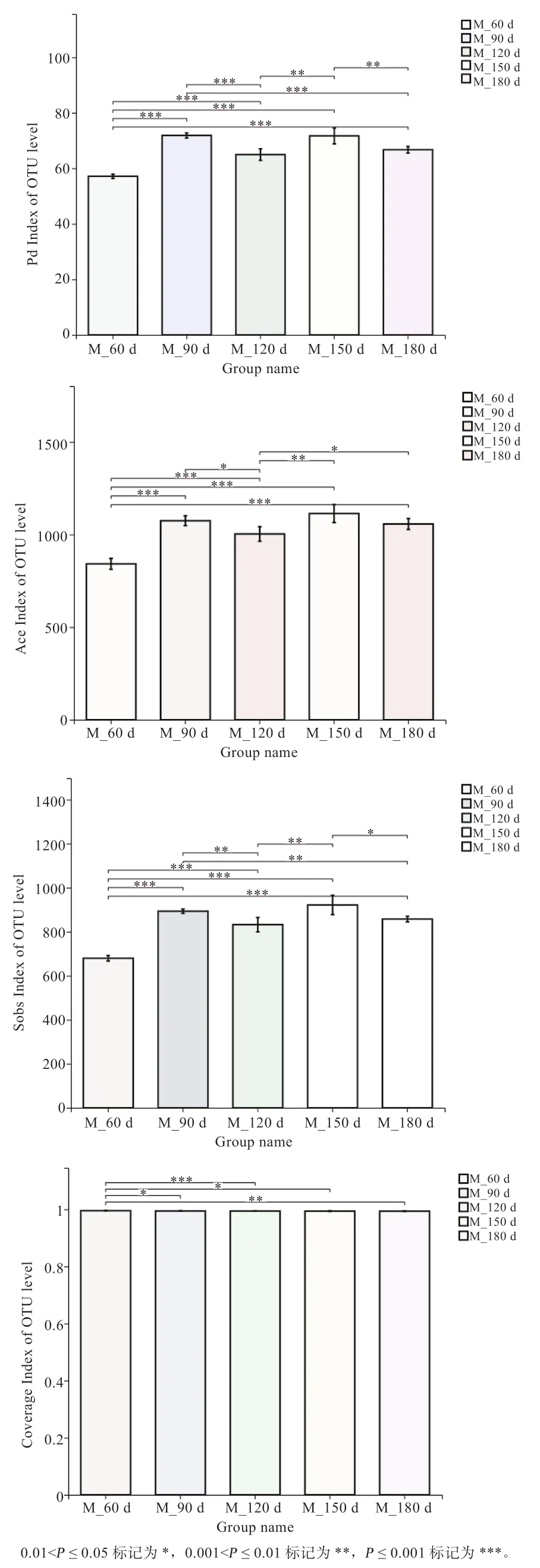
图4 Alpha 多样性指数分析结果图
2.5 肠道微生物不同分类学水平上的群落组成 对所有样本的微生物菌群按照物种丰度进行统计发现,检测到的肠道微生物共涉及22 个门、42 个纲、74 个目、119 个科、321 个属和579 个种,5 组样本主要分布于14 个门,其中厚壁菌门(Firmicutes)、拟杆菌门(Bacteroides)、螺旋体门(Spirochaetae)为主要优势菌门。在门水平上,厚壁菌门丰度呈先升后降的变化趋势,从60 日龄的76.22%,在120 日龄上升到91.84%,在180 日龄又降到72.05%。拟杆菌门丰度呈先降后升的变化趋势,从60日龄的19.47%,在120 日龄降低到5.34%,180 日龄又升到8.46%。螺旋体门(Spirochaetae)所占比例越来越大,从60 日龄的0.21%,180 日龄达到17.82%,采用(One-Way ANOVA)单因素方差分析发现,厚壁菌门(Firmicutes)、拟杆菌门(Bacteroidetes)、螺旋体门 (Spirochaetae)、放线菌门 (Actinobacteria)、无壁菌门(Tenericutes)间有显著差异(表4)。在属水平上,采用(One-Way ANOVA)单因素方差分析发现,共有25 个菌属间存在差异显著,其中60 日龄普氏菌属9(Prevotella_9)的相对丰度最高达10.04%,90 日龄链球菌属(Streptococcus)的相对丰度最高达29.85%,120 日龄梭状芽孢杆菌属(Clostridium_sensu_stricto_1)的相对丰度最高达20.16%,150 日龄Terrisporobacter的相对丰度最高达19.33%,180 日龄螺旋体2 菌属(Treponema_2)的相对丰度最高,高达17.49%(表5)。
2.6 猪肠道微生物与环境因子关联分析 选取丰度值前30 的肠道微生物菌群与血液免疫抗体进行相关性分析(表6),发现IL-2、IL-6、IgG、IgM、IgA 浓度与肠道菌群之间存在显著的相关性(P<0.05)。不同时期肠道菌群与血清指标相关性热图(图5)和相关系数(表7)显示,与血清IL-2 浓度显著相关的有12 个菌种(P<0.05),其中侠义梭状芽孢杆菌属(Clostridium_sensu_stricto_1)与血清IL-2 浓度显著中度正相关,Catenibacterium、Rikenellaceae_RC9_gut_group、norank_f__Bacteroidales_S24-7_group、Roseburia、norank_o__Mollicutes_RF9、unclassified_o__Lactobacillales、Ruminococcaceae_UCG-010、Lachnospiraceae_XPB1014_group、Streptococcus、Ruminococcaceae_UCG-013、Lachnospiraceae_AC2044_group 等10 种菌群 与血清IL-2 浓度显著中等强度负相关。与血清IL-6 浓度显著相关的有14 个菌种,其中unclassified_f__Ruminococc aceae、unclassified_o__Lactobacillales 与血清IL-6 浓度显著中度正相关,unclassified_p__Firmicutes 血清IL-6 浓度显著较强正相关,[Ruminococcus]_torques_group 与血清IL-6 浓度显著较强负相关。与血清IgG浓度显著相关的有12 个菌种,其中Lachnospiraceae_XPB1014_group 与血清IgG 浓度显著中度正相关,Anaerotruncus 与血清IgG 浓度显著较强正相关。IgM 与11 个菌群存在显著相关性,其中norank_f__Bacteroidales_S24-7_group 与血清IgM 浓度显著中等强度正相关,Ruminococcaceae_NK4A214_group 与血清IgM 浓度显著中等强度负相关。IgA 与26 个菌群存在显著相关性,其中Lachnospiraceae_XPB1014_group与血清IgA 浓度显著较强正相关,norank_f__p-2534-18B5_gut_group、Lachnospiraceae_AC2044_group、Prevotellaceae_NK3B31_group、Ruminococcaceae_NK4A214_group、Coprococcus_3、Holdemanella、[Ruminococcus]_torques_group 与血清IgA 浓度显著中等强度正相关,Ruminococcus_1、unclassified_f__Lachnospiraceae、Ruminococcaceae_UCG-010 和Streptococcus 与血清IgA 浓度显著中度负相关。
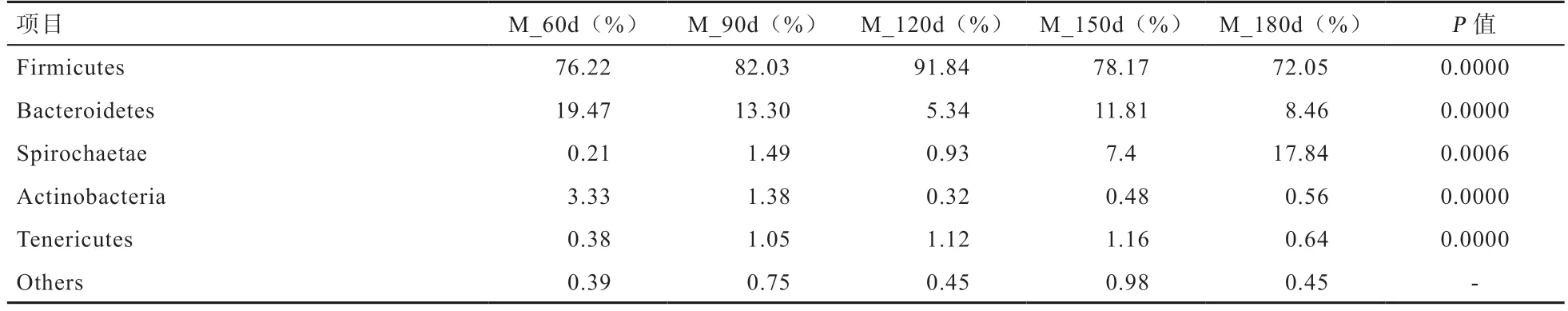
表4 不同时期门水平上主要菌群
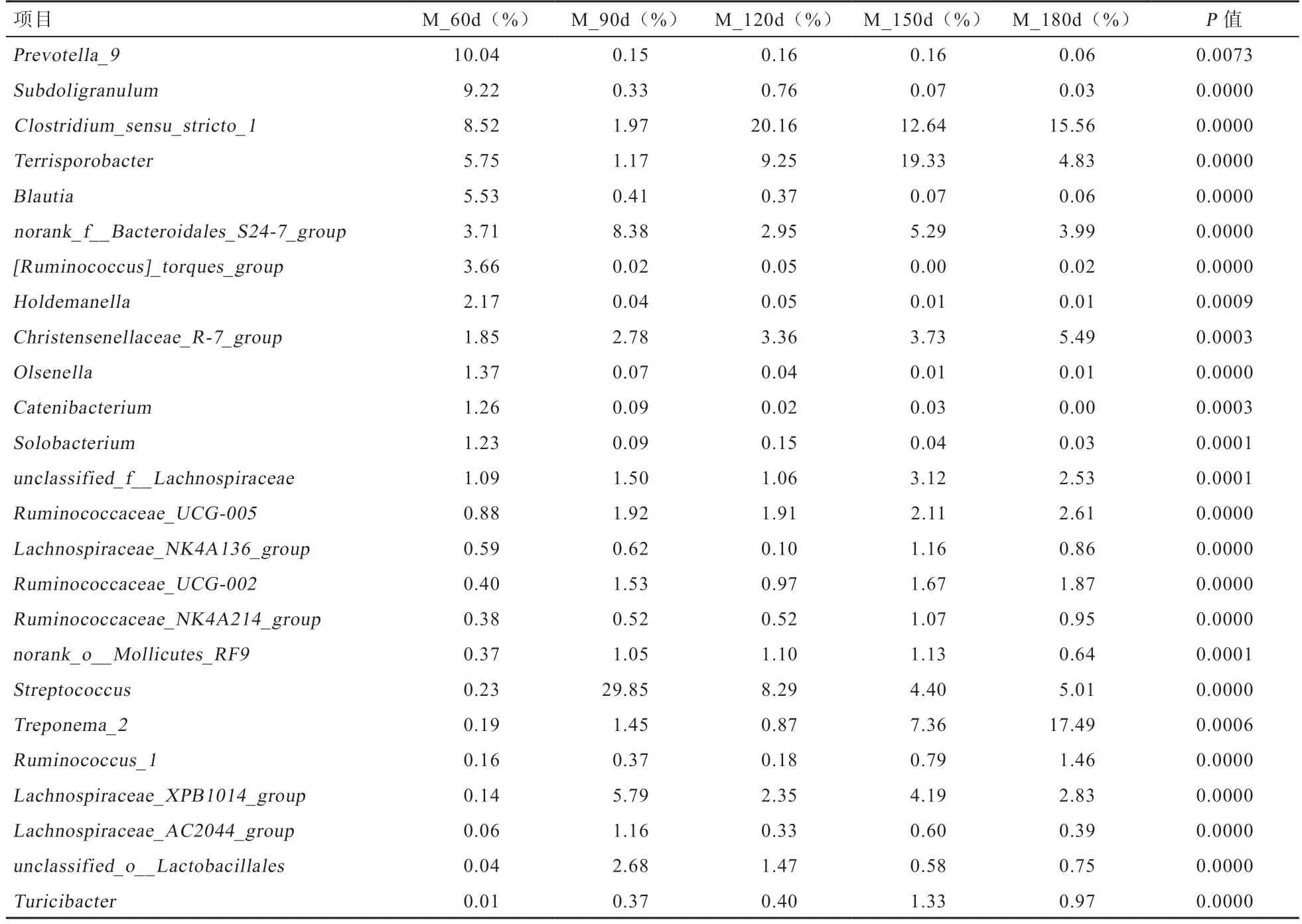
表5 不同时期属水平上主要菌群
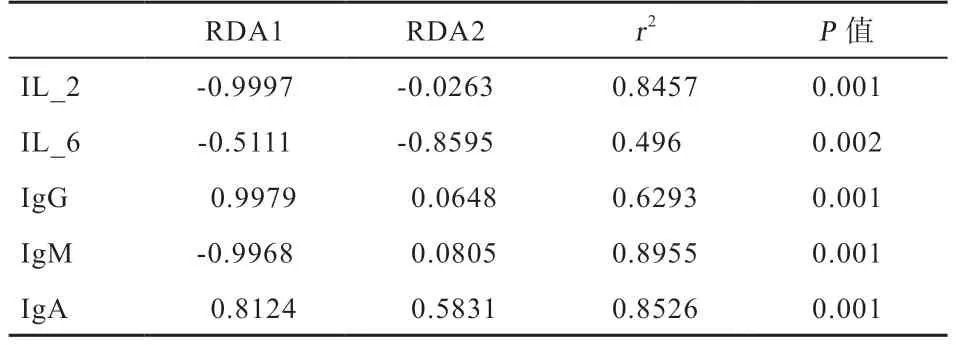
表6 肠道微生物与血液免疫抗体指标RDA 分析结果
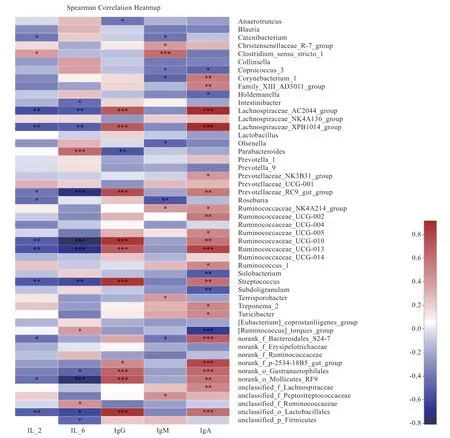
图5 不同时期肠道菌群与血清指标相关性热图

表7 不同时期肠道菌群与血清指标相关系数
3 讨 论
哺乳动物从出生到死亡,肠道微生物菌群是一个动态的变化过程,当外部环境改变后,肠道内的微生态的动态平衡也会随之改变。猪肠道微生物菌群组成和人类相似,主要包括细菌、古菌、真核微生物,大约有1 000 多种,其中优势菌为乳酸菌等厌氧菌占99% 以上,而需氧菌和兼性厌氧菌只占1% 左右[8-9]。在不同的生长阶段,猪肠道的优势菌群主要是厚壁菌门和拟杆菌门,猪肠道优势菌群与自身免疫调控有很大关系,随着日龄的增长和外部环境的改变,肠道中优势菌群也相应变化[10]。本研究中,不同日龄的优势菌群也有所不同,虽然在不同日龄时厚壁菌门(Firmicutes)、拟杆菌门(Bacteroides)和螺旋体门(Spirochaetae)为主要优势菌门,但厚壁菌门、拟杆菌门、螺旋体门中的显著差异菌属差异较大。大肠是消化纤维的主要场所,随着日粮中粗纤维含量增加,Prevotella_9 属于普氏菌属(Prevotella),主要参与多糖的降解和氨基酸的代谢,尤其对纤维素的代谢有重要作用[11]。张冬杰等[12]在研究民猪肠道菌群特征时发现,饲粮中粗纤维的变化能够引起密螺旋体属、疣微菌属、普氏菌属的变化,郭秀兰[13]研究发现密螺旋体菌属能够通过对饲料中多糖和胶质进行发酵,产生短链脂肪酸调节宿主能量平衡,抑制脂肪组织发育的作用,本研究中180 日龄螺旋体菌属(Treponema_2)丰度值最高,可能与饲料中粗纤维的含量较高有关。Liu 等[14]研究玉米麦麸对猪肠道微生物及其细胞因子多样性影响时发现,适当增加粗纤维可以提高盲肠肠道微生物的多样性;王鹏[15]研究发酵玉米秸秆对育肥猪生产性能及盲肠微生物区系的影响时发现,随着纤维含量增加,猪盲肠微生物shannon 指数呈先增加后降低的趋势。张奇等[16]研究发现,日粮中粗纤维增加能够刺激猪回肠中乳酸菌的生长并产生多种抗菌化合物,抑制其他微生物生长,使猪肠道微生物多样性降低。本研究发现,随着饲料中粗纤维的比例越来越高,90 日龄时猪肠道微生物shannon 指数达到最高,随着日龄的增加肠道微生物趋于稳定,猪肠道微生物种的种类数有所减少。
肠道黏膜是猪体内最大的黏膜免疫器官,SIgA 是肠黏膜细胞内分泌量最多的免疫球蛋白,肠道微生物是肠道黏膜免疫的重要调节因子[17]。本研究通过环境因子的关联分析发现,血液中IL_2 浓度与狭义梭状芽孢杆菌属(Clostridium_sensu_stricto_1)呈中度正相关,与拟杆菌属S24-7(norank_f__Bacteroidales_S24-7_group)呈中度负相关,血液中IL_6 浓度与胃瘤球菌属(Ruminococcus)、拟杆菌属、乳杆菌属有较强的相关性,与郭晓红等[18]的研究结果一致。毛螺菌科属在仔猪生长发育过程中对能量代谢、机体免疫能力等方面起着重要作用。Callaway 等[19]研究发现,毛螺菌科属与脂肪能量代谢、丁酸盐代谢有较强的相关性。本研究中发现,毛螺菌属(Lachnospiraceae)的XPB1014、AC2044 与IgA、IgM、IgG 都有显著的相关性。粪球菌属(Coprococcus)对短链脂肪酸的产生具有重要作用,能够阻挡其他有害菌进入到肠道,减少炎症的发生。Min 等[20]发现在不同性别人群中,霍尔德曼氏菌(Holdemanella)会导致脂肪沉积的差异。本研究发现,粪球菌属、霍尔德曼氏菌与血液IgA 浓度都存在中度正相关,说明在猪生长发育的过程中,这些菌种在脂肪代谢过程起着重要作用。仔猪断奶后大肠杆菌、沙门氏菌、葡萄球菌等致病菌在仔猪肠道数量也来越多,引起仔猪肠道功能紊乱的同时也能刺激SIgA 的分泌,拟杆菌属S24-7(Bacteroidales_S24-7)与肠道的炎症有显著的相关性[21]。本研究发现,拟杆菌属S24-7 与IL_2 和IgM间也存在显著的相关。Salminen 等[22]研究表明,当有害菌或病毒引起肠道感染时,肠道内的IgA 要比血液中的IgM、IgG 具有更大的保护作用,在本研究发现,与IgA浓度存在显著相关的菌群多达26 个,说明肠道菌群在抗炎症、能量代谢、提高机体抵抗能力有积极的作用。
4 结 论
本研究以杜× 长× 大为试验对象,探讨仔猪断奶后不同生长阶段和饲料营养水平的变化对猪肠道微生物菌群的影响,发现断奶后随着日龄的不断增加,猪肠道微生物趋于稳定,肠道微生物菌群的种类也有数减少,厚壁菌门(Firmicutes)、拟杆菌门(Bacteroides)、螺旋体门(Spirochaetae)为肠道的主要优势菌门;梭状芽孢杆菌属、乳杆菌属、拟杆菌属、瘤胃球菌属等菌属与血液免疫抗体具有显著相关性,对猪的生长发育有着重要的作用。
