体外偶联UDP-糖基转移酶与蔗糖合成酶高效催化合成莱鲍迪苷A
2021-01-19朱清娟陈美琪梁书利王登刚林影
朱清娟,陈美琪,梁书利,王登刚,林影
(华南理工大学生物科学与工程学院,广东省发酵与酶工程重点实验室,广东广州 510006)
甜菊糖是从甜叶菊叶子中提取和纯化的二萜糖苷,具有高甜度、低热量、对人体无副作用等优点[1],美国食品和药品管理局在 2008年将其列入为一般认为安全(Generally Recognized As Safe,GRAS)的甜味剂[2],其中甜菊苷(Stevioside,ST)和莱鲍迪苷A(Rebaudioside A)是甜菊糖含量最丰富的两种成分,分别占干叶重的5%~10%和2%~4%。甜菊苷ST在甜菊糖中含量最高且甜度是蔗糖的250~300倍,但其具有较严重的苦涩余味,从而限制了其在食品和药品中的消费和应用。然而莱鲍迪苷A(rebaudioside A,RA)除了具有高甜度、低热量(热值仅为蔗糖的1/300)、对人体无副作用等优点外,而且口味纯正,没有后苦味[3]。因此可以作为天然甜味剂在食品和饮料行业广泛应用[4],同时不会因长期食用使血糖浓度增加,可以有效预防肥胖症、糖尿病、高血压、苯丙酮尿症等病症[5]。目前制备高纯度莱鲍迪苷A产品的方法主要包括培育高产莱鲍迪苷A的甜叶菊品种、树脂吸附或重结晶提纯莱鲍迪苷A和环糊精转移酶等酶对甜菊糖进行改性等[6-9],但都存在工艺繁琐、成本高、效果不佳等缺点。已经有研究发现在糖基转移酶(Glycosyltransferases,GTs)类中存在 UDP-糖基转移酶UGT76G1能特异性地催化甜菊苷通过一步糖基化反应合成莱鲍迪苷 A[10]。然而,甜叶菊中天然UGT76G1含量甚微,分离纯化成本高,难以获取大量的酶蛋白,并且糖基转移反应中需要昂贵的尿苷二磷酸葡萄糖(UDPG)作为糖基供体[11,12],必然导致RA苷酶促合成应用的经济性差,解决糖基化反应过程中糖基转移酶的制备及底物UDPG昂贵的问题是提高RA生产效益的主要方法。
蔗糖合成酶(Sucrose synthase,SuSy)催化可逆的葡萄糖基转移反应,通过蔗糖合成酶催化蔗糖和UDP产生UDP-葡萄糖,有效地为UDP-糖基转移酶提供了UDP-糖的需求[13,14]。在偶联蔗糖合成酶和UDP-糖基转移酶构建的级反应体系,产生 UDP循环,实现 UDP-葡萄糖的再生已成为糖基化生产权宜的供体[15],已知的两类蔗糖合成酶,分别来源于植物和细菌,通常来源于细菌的蔗糖合成酶对ADP有高的亲和力,而对UDP亲和力相对低,植物蔗糖酶对UDP的亲和力高[16]。最近的研究发现选择细菌来源的蔗糖合成酶耐热稳定性、比酶活和重组酶表达水平明显高于植物来源的酶,因此其工业化应用前景引起了人们极大的兴趣。目前已有研究者在毕赤酵母中共表达 UDP-糖基转移酶UGT76G1和拟南芥来源的AtSUS这两个酶基因在细胞体内全细胞催化ST转化为RA,其中唐小雁等以10 mg/mL的ST为底物合成为7.46 mg/mL的RA,RA的收率为74.60%[17]。同时也有研究者在大肠杆菌宿主菌中结合来自 rebaudiana的重组 UDP-葡萄糖基转移酶 UGT76G1和来自拟南芥的蔗糖合酶AtSUS1的活性,外源添加2.40 mM甜菊糖苷,7.20 mM蔗糖和0.006 mM UDP反应30 h,莱鲍迪苷A的收率为78.21%[18],此前报道的酿酒酵母中利用UDP-糖基转移酶UGT76G1将ST转化为RA,其中刘欢等以1 g/L的ST为底物合成267.89 mg/L[19]。
本研究分别构建了表达高活性 UDP-糖基转移酶UGT76G1和绿豆来源的蔗糖合成酶mbSUS的重组毕赤酵母菌株。通过发酵产酶,制备 UDP-糖基转移酶UGT76G1和蔗糖合成酶mbSUS,建立级联酶催化反应体系合成RA,实现UDPG在生物催化反应中的循环,如图1所示。此外,进一步探究生物催化体系中,两种酶不同的酶活配比量对RA合成的影响,确定了此级联反应中的限速酶和最适酶活配比,为RA酶法生物合成及其产业化应用提供技术支持。

图1 UGT76G1和蔗糖合成酶级联反应催化体系Fig.1 Process of catalytic synthesis of RA
1 材料与方法
1.1 材料
1.1.1 菌株与质粒
宿主菌菌株(E.coli Top)10F”,(Pichia pastoris)GS115以及质粒pPICZA由本实验室保存。毕赤酵母表达质粒 pPIC9K,购买自 Invitrogen公司。质粒pUC57-mbSUS和质粒pUC57-UGT76G1,带已通过毕赤酵母密码子优化后的基因mbSUS和UGT76G1。这两个优化基因由南京金斯瑞生物科技有限公司全基因合成。
1.1.2 工具酶与试剂
限制性内切酶EcoRI、NotI、SalI和MssI购自宝生物工程(大连)有限公司;高保真、高效率扩增目的基因片段的KOD-Plus-Neo DNA聚合酶,购买自东洋纺(上海)生物科技有限公司。质粒小量制备试剂盒和琼脂糖凝胶DNA回收试剂盒购自上海捷瑞生物工程有限公司;PCR纯化试剂盒购Magen(美基)生物有限公司;甜菊苷、莱鲍迪苷A由曲阜海根甜菊制品有限公司提供;UDP,UDPG购买自上海源叶生物科技有限公司。
1.1.3 培养基
LBL平板培养基:1%胰蛋白胨,0.5%酵母抽提物,0.5% NaCl,1.5%的琼脂粉,根据需要添加zeocin使终浓度为 100 mg/mL。YPD平板培养基:1.34%YNB;0.4 mg/L生物素;2%葡萄糖,1.5%的琼脂粉。YPD(含 Zeocin)平板:1%酵母粉,2%蛋白胨,2%葡萄糖,2%琼脂粉,100 μg/mL Zeocin,MD 平板:1.34%YNB,2%葡萄糖,2%琼脂粉,(4×10-5)% Biotin. BMGY培养基:1.34% YNB,1%酵母提取物,2%蛋白胨,10%PBS(pH 6.0),1%甘油。BMMY培养基:1.34% YNB,1%酵母提取物,2%蛋白胨,10% PBS(pH 6.0)。
1.2 方法
1.2.1 蔗糖合成酶目的基因的优化及合成
在 NCBI数据库中获得绿豆来源的蔗糖合成酶(Sequence ID:BAA01108.1)。将该基因氨基酸序列送往南京金斯瑞生物科技有限公司,经毕赤酵母密码子优化而获得的碱基序列,全基因合成后克隆至质粒pUC57,得到含有蔗糖合成酶编码基因的重组质粒pUC57-mbSUS。
1.2.2 蔗糖合成酶mbSUS表达载体的构建
在mbSUS基因的两端引入EcoRI和NotI限制性酶切位点,如下所示:mbSUS-F:5'-ACGAGGAATTCA TGGCTACTGACAGATT-3'(EcoRI);mbSUS-R:5'-GG CGGCCGCCTCAACA GCCAATGGAACA-3'(NotI)。以含有编码蔗糖合成酶并经毕赤酵母密码子优化的基因mbSUS的载体pUC57-mbSUS为模板,进行PCR扩增。将 PCR产物用试剂盒纯化,用限制性内切酶EcoRI和NotI对PCR回收产物及质粒pPICZA分别进行双酶切,纯化回收后用T4连接酶进行连接反应,连接产物转化大肠杆菌E.coli感受态细胞,得到表达载体pPICZA-mbSUS。
1.2.3 糖基转移酶目的基因的优化及合成
在NCBI数据库中获得不含其自身信号肽的甜叶菊UGT76G1氨基酸序列(Sequence ID:AGL95113.1)。将该氨基酸序列送往南京金斯瑞生物科技有限公司,经毕赤酵母密码子优化而获得的碱基序列,全基因合成后放入质粒 pUC57-UGT76G1。糖基转移酶UGT76G1目的基因的优化及合成。
1.2.4 糖基转移酶UGT76G1表达载体的构建
在UGT76G1基因的两端引入限制性酶切位点,如下所示:UGT76G1-F:5'-GGATCCATAAGACCGAG ACTACCGTCAGAAGA-3'(BamH I);UGT76G1-R:5'-GCGGCCGCTATGAAACAAGGGACTCCAAAGATT-3'(NotI)。以含有编码蔗糖合成酶并经毕赤酵母密码子优化的基因UGT76G1的载体pUC57-UGT76G1为模板,进行PCR扩增。将PCR产物用试剂盒纯化,用限制性内切酶BamH I和NotI对PCR回收产物及质粒pPIC9K分别进行双酶切,纯化回收后用T4连接酶进行连接反应,连接产物转化大肠杆菌E.coli感受态细胞,得到表达载体pPIC9K-UGT76G1。
1.2.5 重组毕赤酵母菌株的构建
重组质粒pPICZA-mbSUS用限制性内切酶MssI线性化,电击转化毕赤酵母GS115/pPIC9K感受态中,涂布于YPDZ平板,于30 ℃恒温培养2~4 d,挑取博来霉素抗性重组转化子,微波裂解菌体获得基因组。以基因组为模板,进行PCR鉴定筛选得到表达蔗糖合成酶的重组毕赤酵母菌株。重组质粒 pPIC9KUGT76G1用限制性内切酶SalI线性化,电击转化毕赤酵母GS115感受态中,涂布于MD平板,于30 ℃恒温培养 2~4 d,挑取重组转化子,微波裂解菌体获得基因组。以基因组为模板,进行PCR鉴定筛选得到重组工程菌GS115/pPIC9K-UGT76G1。分别取这两株毕赤酵母重组工程菌接种到10 mL BMGY液体培养基(50 mL的锥形瓶)中,30 ℃恒温摇床250 r/min振荡培养16~18 h,至OD600达到4~6。离心后转至25 mL BMMY培养基(250 mL的锥形瓶,控制起始OD值约为1.0),30 ℃恒温摇床250 r/min振荡培养。每隔24 h取250 µL诱导培养液,用于分析重组菌的生长情况。每次取样完毕后,补加 1%甲醇,连续诱导发酵120 h。
1.2.6 糖基转移酶和蔗糖合成酶酶活的测定
取200 µL诱导培养液于干净的2 mL离心管中,控制菌体OD600=60,用蒸馏水洗涤三次之后,弃上清。加入1 mL蒸馏水转移至装有0.73 g破碎珠的破碎管中,再加入30 µL 1%的蛋白酶抑制剂,置于破碎仪中进行破碎,800 r/min破碎1 min后在冰上冰冻1 min,反复破碎8次之后在4 ℃离心机中12000 r/min离心10 min,取上清到2 mL干净的离心管中,放置在冰上保存备用。糖基转移酶UGT76G1与反应底物混合,200 µL反应体系终浓度为pH 7.0的50 mM磷酸钾缓冲溶液,10 mM MgCl2,10 mM ST,2 mM UDPG,45 ℃摇床200 r/min反应20 min。蔗糖合成酶mbSUS与反应底物混合,200 µL反应体系终浓度为 pH 7.0的50 mM磷酸钾缓冲溶液,10 mM MgCl2,10 mM 蔗糖,2 mM UDP,45 ℃摇床200 r/min反应20 min。反应结束后加入200 µL 0.2 mM的磷酸溶液,静置5 min使酶失活,再加入200 µL的0.2 mM的氢氧化钠溶液中和pH值。终止反应结束后加入400 µL的蒸馏水稀释,涡旋振荡,14000 r/min离心2 min,取上层液体用0.22 µm膜过滤后用于HPLC分析。
色谱检测条件:pHenomenex Gemini 5u C18柱(250 mM×4.6 mM),流动相为0.20 mol/L的磷酸盐缓冲液(17.90 g/L磷酸氢二钠缓冲液和6.80 g/L的磷酸二氢钾缓冲液)和1.61 g/L的四丁基溴化铵的纯水相,柱温35 ℃,流速1 mL/min,进样量为10 µL,紫外检测波长262 nm。使用UDPG标准品(100%)和UDP(99%)通过外标法进行定量。UDPG和UDP浓度的线性范围为0.1~2 g/L。
得到UDP和UDPG色谱峰面积,根据标准曲线换算得到反应生成UDP或者消耗的UDP量,通过一下酶活计算公式计算蔗糖合成酶 mbSUS和糖基转移酶UGT76G1的酶活。
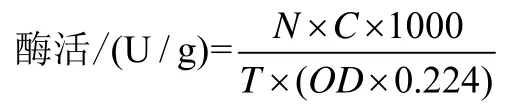
注:酶活力单位定义为在上述反应条件下,1 min催化形成1 mol UDPG所需要的酶量为1个活力单位,其中N:反应液后期处理的稀释倍数,C:根据标准曲线换算得到的生成UDPG或UDP的量,T:反应时间。
1.2.7 不同酶量配比生物催化合成RA
用ST为底物分析新构建的两株重组酵母菌中的酶以不同酶活比例混合条件生物催化合成 RA。甲醇诱导摇瓶发酵120 h后离心收集两株菌体,用100 mM磷酸钾缓冲液(pH 7.0)洗涤两次。参照1.2.6的破碎方法破碎之后收集上清液,测定酶活。
控制体系中UGT76G1(K)的酶活为10 U,不同体系中mbSUS(S)的酶活为10、20、40、60 U,使体系中两个酶的酶活配比为 K1+S1、K1+S2、K1+S4、K1+S6按照不同的酶活比例添加到200 µL反应体系中,混合体系中各物质终浓度分别为pH 7.0的50 mM磷酸钾缓冲溶液,3 mM MgCl2,10 mmM ST,0.6 mM UDP,50 mM蔗糖。将混合物在45 ℃下反应3 h后终止反应。反应体系用蒸馏水稀释五倍,经0.22 µm膜过滤后用于HPLC分析。
控制体系中mbSUS(S)的酶活为10 U,不同体系中UGT76G1(K)的酶活为10、20、40、60 U,使体系中两个酶的酶活配比为 K1+S1、K2+S1、K4+S1、K6+S1。按照不同的酶活比例添加到200 µL反应体系中,混合体系中各物质终浓度分别为pH 7.0的50 mM磷酸钾缓冲溶液、3 mM MgCl2、10 mM ST、0.6 mM UDP、50 mM蔗糖。将混合物在45 ℃下反应3 h后终止反应。反应体系用蒸馏水稀释五倍,经0.22 µm膜过滤后用于HPLC分析。
控制不同体系中 mbSUS(S)和 UGT76G1(K)酶活配比,使其中配比分别为K1+S1(K=10 U,S=10 U)、K2+S2(K=20 U,S=20 U)、K4+S4(K=40 U,S=40 U)、K6+S6(K=60 U,S=60 U)。按照不同的酶活比例添加到200 µL反应体系中,混合体系中各物质终浓度分别为pH 7.0的50 mM磷酸钾缓冲溶液、3 mM MgCl2、10 mM ST、0.6 mM UDP、50 mM蔗糖。将混合物在45 ℃下反应3 h后终止反应。反应体系用蒸馏水稀释五倍,经0.22 µm膜过滤后用于HPLC分析。
1.2.8 数据统计分析
每个重组酵母菌株重复发酵三次,每次酶活测定以及反应合成RA设置三个平行,数据值为平行样品的平均值,用误差线表示标准偏差。
2 结果与讨论
2.1 糖基化转移酶UGT76G1和蔗糖合成酶毕赤酵母表达质粒的构建
以质粒pUC57-mbSUS为模板,PCR扩增得到约2400 bp的mbSUS基因片段,经限制性内切酶EcoRI和NotI双酶切后,连接载体片段构建pPICZA- mbSUS重组质粒。重组质粒经限制性内切酶EcoRI和NotI双酶切并用凝胶电泳鉴定。如图2a所示,双酶切得到两条带分别为3279 bp和2426 bp,与预期的目的片段大小一致,经DNA测序后无碱基突变,结果正确,重组质粒成功构建。重组质粒pPICZA- mbSUS经限制性内切酶MssI线性化后电转化毕赤酵母GS115/pPIC9K感受态中。YPDZ平板初筛得到的重组转化子,微波裂解粗提基因组经PCR凝胶电泳鉴定。如图2b所示,在2700 bp左右扩增出一条特异性条带,与预期的目的片段大小一致,表明pPICZA-mbSUS已成功整合到GS115基因组上。

图2 重组质粒pPICZA-mbSUS的双酶切鉴定及转化子PCR鉴定Fig.2 Dual-enzyme identification of transformant pPICZA-mbSUS and PCR identification of transformants
以质粒pUC57-UGT76G1为模板,PCR扩增得到约1377 bp的UGT76G1基因片段,经限制性内切酶BamH I和NotI双酶切后,连接载体片段构建pPIC9K-UGT76G1重组质粒。重组质粒经限制性内切酶BamH I和NotI双酶切并用凝胶电泳鉴定。如图3a所示,双酶切得到两条带分别约为8979 bp和1376 bp,与预期的目的片段大小一致,经DNA测序后无碱基突变,结果正确,重组质粒成功构建。重组质粒pPIC9K-UGT76G1经限制性内切酶SalI线性化后电转化毕赤酵母GS115感受态中。MD平板初筛得到的重组转化子,微波裂解粗提基因组经PCR凝胶电泳鉴定。如图3b所示,在1376 bp左右扩增出一条特异性条带,与预期的目的片段大小一致,表明 UGT76G1已成功整合到GS115基因组上。

图3 重组质粒pPIC9K-UGT76G1的双酶切鉴定及转化子PCR鉴定Fig.3 Dual-enzyme identification of transformant pPIC9K-UGT76G1 and PCR identification of transformants
2.2 糖基转移酶UGT76G1和蔗糖合成酶毕赤酵母高效表达体系的构建

图4 重组毕赤酵母菌株生长曲线Fig.4 Recombinant Pichia pastoris growth curve
重组菌株经甲醇诱导120 h后,生长情况正常,如图4所示。从图上看出,随着发酵的进行,两株重组菌株的菌体数量都逐渐提高,甲醇诱导到120 h时生长趋于稳定。两株重组菌株生长情况相似,与对照菌株的生长情况相比较,两株重组毕赤酵母菌株的生长情况均受到了一定影响。
收集经甲醇诱导120 h后的菌液,蒸馏水洗涤3遍后进行破碎处理,提取上清液按照1.2.6的方法测定酶活。根据酶活公式计算重组菌株中糖基转移酶UGT76G1和蔗糖合成酶mbSUS的酶活,所得酶活如图5所示,在反应条件下,糖基转移酶UGT76G1的酶活约为141.22 U/g,蔗糖合成酶mbSUS的酶活约为121.39 U/g。

图5 重组毕赤酵母菌株UGT76G1和蔗糖合成酶产酶分析Fig.5 Enzyme activity of recombinant Pichia pastoris strains
2.3 糖基转移酶UGT76G1和蔗糖合成酶级联反应催化体系构建及莱鲍迪苷A合成
在生物催化反应中,添加 U(UGT76G1):U(mbSUS)=1:1的条件下反应3 h,RA产量为4.96 mM,固定体系中UGT76G1的酶活为10 U,不同体系中mbSUS酶活分别为10、20、40、60 U,使体系中两个酶的酶活配比为K1+S1,K1+S2,K1+S4,K1+S6,如图6 a所示,随着蔗糖合成酶mbSUS酶活的增加,RA产量增加。在添加U(UGT76G1):U(mbSUS)=1:6的条件下反应3 h,RA产量为5.81 mM,与U(UGT76G1):U(mbSUS)=1:1酶活比例条件下RA产量相比提高了17.14%。在提高mbSUS的酶活情况下,蔗糖和UDP作为底物充足的条件下,积累了大量的UDPG,而糖基转移酶UGT76G1的量是固定的,继续增加mbSUS,UGT的转化速度可能跟不上 mbSUS的转化速度,因此不能大量催化合成RA。
固定体系中mbSUS的酶活为10 U,不同体系中UGT76G1酶活分别为10、20、40、60 U,使体系中两个酶的酶活配比为K1+S1,K2+S1,K4+S1,K6+S1,如图6b所示,随着糖基转移酶UGT76G1酶活的增加,RA产量有明显的增长。当UGT76G1的酶活增加到60 U时,与添加40 U的糖基转移酶相比,RA产量仅提高7.04%。说明此时,体系内UGT与mbSUS的循环速率可能达到最佳,级联反应获得最优平衡。添加60 U糖基转移酶 UGT76G1和 10 U蔗糖合成酶mbSUS反应 3 h,RA产量为 8.20 mM,与U(UGT76G1):U(mbSUS)=1:1酶活比例催化相比,RA产量提高了65.32%,在此生物催化级联反应过程中,糖基转移酶的量增加到一定程度,由于缺少蔗糖合成酶催化蔗糖生成果糖提供UDPG作为底物催化甜菊糖苷生物合成大量的莱鲍迪苷A。
在催化反应过程之中,同时增加 UGT76G1和mbSUS的酶量,研究其对合成RA的影响。如图6c所示,糖基转移酶 UGT76G1和蔗糖合成酶 mbSUS的酶量同时增加,RA的产量有少量增加,糖基转移酶mbSUS能可逆催化蔗糖和UDP生成UDPG,提高蔗糖合成酶的酶活,可以大量积累UDPG。在催化反应体系中,UDPG作为底物可以通过糖基转移酶UGT76G1催化甜菊糖苷生成莱鲍迪苷 A,随着糖基转移酶UGT76G1和蔗糖合成酶mbSUS酶活的同时增加,RA产量提高,在添加U(UGT76G1):U(mbSUS)=6:6的条件下反应 3 h,RA 产量为 6.38±1.03 mM,与U(UGT76G1):U(mbSUS)=1:1酶活比例反应相比,RA产量提高了28.63%。
在对上述不同反应条件进行比较后,得到糖基转移酶UGT76G1和蔗糖合成酶mbSUS这两个酶在不同酶量比例反应生成 RA的最适酶活比例为U(UGT76G1):U(mbSUS)=6:1,反应3h RA产量为8.20±0.11 mM,RA的产率达到82.91%。如图6d所示,最适的反应条件为单独提高糖基转移酶 UGT676G1的酶活到60 U,添加蔗糖合成酶mbSUS的酶活为10 U,在此酶活比例条件下既避免蔗糖合成酶 mbSUS过量,UGT的转化速度跟不上mbSUS的转化速度,同时也提供适量的糖基转移酶UGT76G1进行催化糖基转移化反应,UGT作为级联反应中的限速酶,提高其比例,能有效推动循环,提高RA产量。


图6 UGT76G1和蔗糖合成酶不同比例组合催化合成莱鲍迪苷A效率比较Fig.6 Comparison of the efficiency of catalytic synthesis of rebaudioside A with different ratios of UGT76G1 and sucrose synthase
3 结论
3.1 莱鲍迪苷A作为甜菊糖苷中甜味高、口感好的成分,获取高纯度莱鲍迪苷A的产品是甜菊糖产品的一个重要发展方向。本研究利用毕赤酵母重组高效表达糖基转移酶UGT76G1和蔗糖合成酶mbSUS,建立了双酶级联生物催化反应体系,优化了添加两个酶的最佳酶量配比为U(UGT76G1):U(mbSUS)=6:1,反应3 h RA产量为8.20±0.11 mM,RA的产率达到82.91%,较添加U(UGT76G1):U(mbSUS)=1:1酶活比例条件催化,RA产量提高了1.65倍。与此前唐毕赤酵母重组菌株相比,RA产量是其全细胞催化反应所得RA产率的1.10倍,生物催化反应时间缩短了5倍,与在大肠杆菌中进行糖基转移酶 UGT76G1和拟南芥来源的蔗糖合成酶AtSUS纯酶催化RA相比,本研究催化所得RA的产率是其催化反应所得RA产率的1.22倍,且反应时间缩短了10倍。
3.2 本研究通过添加糖基转移酶UGT76G1与蔗糖合成酶mbSUS,可以在生物催化过程中实现级联反应,实现催化反应过程中UDPG的循环,在提高了RA产量的基础上,极大的减少了催化反应时间,提高了RA的生产效率。此外本研究发现在此级联反应中糖基转移酶UGT76G1作为限速酶在一定程度上影响了 RA的合成,后续研究可以通过提高基因剂量实现外源蛋白在毕赤酵母系统的高效表达。
