Signatures within esophageal microbiota with progression of esophageal squamous cell carcinoma
2021-01-18MinjuanLiDantongShaoJiachenZhouJianhuaGuJunjieQinWenChenWenqiangWei
Minjuan Li ,Dantong Shao ,Jiachen Zhou ,Jianhua Gu ,Junjie Qin ,Wen Chen ,Wenqiang Wei
1National Central Cancer Registry,National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital,Chinese Academy of Medical Sciences and Peking Union Medical College,Beijing 100021,China;2 Department of Epidemiology and Biostatistics,School of Public Health,Xi’an Jiaotong University Health Science Center,Xi’an 710061,China;3Promegene Translational Research Institute,Shenzhen 518000,China;4Department of Cancer Epidemiology,National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital,Chinese Academy of Medical Sciences and Peking Union Medical College,Beijing 100021,China
Abstract Objective:Esophageal squamous cell carcinoma (ESCC) is one of the dominant malignances worldwide,but currently there is less focus on the microbiota with ESCC and its precancerous lesions.Methods:Paired esophageal biopsy and swab specimens were obtained from 236 participants in Linzhou,China.Data from 16S ribosomal RNA gene sequencing were processed using quantitative insights into microbial ecology(QIIME2) and R Studio to evaluate differences.The Wilcoxon rank sum test and Kruskal-Wallis rank sum test were used to compare diversity and characteristic genera by specimens and participant groups.Ordinal logistic regression model was used to build microbiol prediction model.Results:Microbial diversity was similar between biopsy and swab specimens,including operational taxonomic unit (OTU) numbers and Shannon index.There were variations and similarities of esophageal microbiota among different pathological characteristics of ESCC.Top 10 relative abundance genera in all groups include Streptococcus,Prevotella,Veillonella,Actinobacillus,Haemophilus,Neisseria,Alloprevotella,Rothia,Gemella and Porphyromonas.Genus Streptococcus,Haemophilus,Neisseria and Porphyromonas showed significantly difference in disease groups when compared to normal control,whereas Streptococcus showed an increasing tendency with the progression of ESCC and others showed a decreasing tendency.About models based on all combinations of characteristic genera,only taken Streptococcus and Neisseria into model,the prediction performance was the ideal one,of which the area under the curve (AUC) was 0.738.Conclusions:Esophageal biopsy and swab specimens could yield similar microbial characterization.The combination of Streptococcus and Neisseria has the potential to predict the progression of ESCC,which is needed to confirm by large-scale,prospective cohort studies.
Keywords:Esophageal squamous cell carcinoma;precancerous lesions;16S rRNA;Streptococcus;Neisseria
Introduction
Esophageal cancer (EC) is one of the most usual malignancies in the world,ranking as the ninth and fifth leading causes of global cancer related morbidity and death(1). The two main types of EC are esophageal adenocarcinoma (EAC) and esophageal squamous cell carcinoma (ESCC),and the latter is the predominant histologic subtype in China.The overall 5-year survival rate of ESCC is about 30.3%,which is mostly due to late stage at diagnosis and low early diagnosis rate (2).China has about half of all ESCC cases on earth for its large population of China and high rates.Given the heavy ESCC burden in China and worldwide,it is necessary to develop and advance early detection,early diagnosis and early treatment.
The exact cause of ESCC,which is still unclear,is the powerful basis of effective prevention.As the emerging of microbiome project and high-throughput sequencing method,there are more and more studies about the role of human microbiome in health and disease,which reveals the microbial characterization in carcinogenesis (3).Millions of microorganisms colonizing the gastrointestinal tract are vital to the health of the host and play a role in development,nutrition,immunization and multi-biological process (4-6).Once the microbiota get into unbalanced,alteration may occur to the microenvironment and then lead to carcinoma (7).Several studies (8-10) have showed the microbial diversity and composition of upper gastrointestinal tract (UGI),showing a possible association between the microbiota and carcinoma.And the richness,diversity,and exact composition of microbiota in different organs might contribute to the development of ESCC.However,compared with gastric diseases,less attention was paid to esophageal microbiota and ESCC let alone precancerous lesions.
To gradually explore the microbial signature of esophagus with the progression of ESCC,two key points are worthy of consideration.One point is the sampling method.About ESCC or precancerous lesions,esophageal biopsy is invasive but pathological diagnosis on esophageal biopsy is the “Gold standard”.And mucosal swab sampling seems to be less invasive and with higher performance but not “Gold standard” (11,12).Consequently,biopsy (13,14)and swab specimens (15-17) are the main collection methods for esophageal tissues.However,more appropriate sampling methods are still to be approved for various esophageal diseases.Another point is that diverse and dynamic microbial characterization concerning the progression of ESCC is still unclear,including precancerous disease,precancerous lesions and ESCC.To answer the above two points,paired esophageal biopsy and swab specimens were collected from normal participants and participants with precancerous diseases,precancerous lesions and ESCC.
Materials and methods
Study participants
This study was based on the endoscopy screening of UGI cancer project in China.We retrospectively recruited 236 participants aged 40-69 years with pathologic diagnosis of normal,esophagitis,low-grade intraepithelial neoplasia(LGIN),high-grade intraepithelial neoplasia (HGIN) and ESCC at the Linzhou Cancer Hospital in Henan province in China.All participants were local residents who were screened for UGI cancer in August to September 2018 or January to March 2019.All participants in this study were clearly informed and signed written informed consent.This study was approved and overseen by the Institutional Review Board of the Cancer Hospital of Chinese Academy of Medical Sciences.Participant information,including participant’s sex,age,smoking and alcohol drinking status,dietary habit,history of digestive system disease and oral condition,was collected by trained staff.
Sample collection
Paired biopsy and swab specimens from each participant were obtained from 236 participants under upper gastroenterology endoscopic examination during screening of UGI cancer.The mucosal specimens were collected using sterile swabs (Puritan,sterile polyester tipped applicators) prior to biopsies to prevent contamination of mucosal specimens with blood.If there were lesions,5 looping brushes were taken at the lesion,or else 5 looping brushes were taken at the middle esophagus.After swab specimens were collected,the swab head was cut off into sterile tubes (Cryovial,cryogenic tube 3.0 mL) including 1.5 mL of cell preserving fluid (Hologic,ThinPrep,PreservCyt Solution,San Diego,USA).Biopsy specimens at the brush sampling site were macro-dissected by an experienced endoscopy doctor with sterile forceps under strict procedures,which were then placed into a sterile tube.All paired specimens were stored at -80 °C immediately after sampling and shipped to the laboratory on dry ice.
DNA extraction,amplification and sequencing
Esophageal biopsy and swab specimens were macrocollected and used for DNA extraction by the QIAGEN PowerSoil DNA Isolation Kit 12888100 (QIAGEN,Germany).The extracted DNA was stored at -80 °C in Tris-EDTA buffer solution prior to other processes.To control reagent contamination,we included water as a negative control without DNA template during specimen processing.The V4 region of the 16S ribosomal RNA(rRNA) gene was amplified using the universal bacterial primer set to generate amplicons.The primer pairs for DNA amplification covering variable V4 region were generated using the following primers: 515 F (5’-GTGYCAG-CMGCCGCGGTAA-3’) and 806 R (5’-GGACTACNVGG-GTWTCTAAT-3’) (18,19),incorporating the barcode sequences for 16S rRNA gene.Polymerase chain reaction (PCR) mixtures contained 1 μL of forward and reverse primer (10 μmol/L),1 μL of template DNA,4 μL of deoxyribonucleoside-triphosphates (dNTPs)(2.5 mmol/L),5 μL of 10× EasyPfu Buffer,1 μL of EasyPfu DNA Polymerase (2.5 U/μL) and 1 μL of double distilled water into a 50 μ L total reaction volume.The PCR thermal cycling consisted of an initial denaturation step at 95 °C for 5 min,followed by 30 cycles of denaturation at 94 °C for 30 s,annealing at 60 °C for 30 s,and extension at 72 °C for 40 s,with a final extension step at 72 °C for 4 min.Amplicons from each specimen were run on an agarose gel to ensure consistent sequencing length.Expected band size for 515F to 806R is 300-350 bp.Amplicon quantification was performed using the Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific/Invitrogen catalog no.Q32854,Waltham,USA).The amplicon library for high-throughput sequencing was combined at equal concentrations and volumes and subsequently quantified (KAPA Library Quantification Kit KK4824,USA).Using Illumina V4 chemistry and pairedend 2×150 bp reads,sequencing was performed on the Illumina MiniSeq platform.All sequencing was performed in a single MiniSeq run.Original sequence data processing was performed by the Illumina MiniSeq Reporter to remove adapter and primer sequences and then sequence data were exported in the FASTQ format.
Sequence processing and taxonomic classification
Except one biopsy in esophagitis and HGIN, 470 specimens of the total 472 specimens collected were successfully amplified and sequenced.Sequencing data were processed using the Quantitative Insights into Microbial Ecology (QIIME2,https://qiime2.org/) platform(20).Raw sequences were under strict quality control and feature table construction using the Divisive Amplicon Denoising Algorithm 2 (DADA2) algorithm (21).Possible phiX reads and chimeric sequences were removed,and the remaining reads were truncated from 0 to 140 bp (for both forward and reverse reads) to avoid including sequencing errors at the ends of the reads.Paired-end reads were matched at a maximum mismatch parameter of six bases,which indicates a minimum similarity threshold of 90% for the overlap of the forward and reverse reads.The representative sequences (named “features” in QIIME2 nomenclature) were then generated by removing the redundant and low occurrence (n<5 within all samples)sequences.Data from all samples were rarefied to 1,000 reads for both diversity and relative abundance to avoid bias due to different sampling depths.The taxonomic assignment of the sequence variants (99% similarity) was assigned using the trained Naive Bayes classifier [trained on the Greengenes 13_8 (22)] through the q2-feature-classifier plugin,and the taxonomic composition at the phylum and genus level was generated based on operational taxonomic units (OTU) annotation.
Statistical analysis
All statistical analyses were performed with software program R Studio (Version 1.1.456;R Foundation for Statistical Computing,Vienna,Austria).Differences in observed OTUs and Shannon index were analyzed between paired esophageal biopsy and swab specimens in each participant group by the Wilcoxon rank-sum test.And the observed OTUs and Shannon index among five participant groups were compared by the Kruskal-Wallis test in esophageal biopsy and swab specimens, respectively.Principal coordinates analysis (PCoA) and the Adonis test were used to find discrepancy among the independent β diversity matrices.The relative abundance was calculated at the phylum and genus level for each participant group in each type of specimens.High relative abundance (≥0.01)genera were compared between normal and other four participant groups by the Wilcoxon rank-sum test and were compared among five participant groups by the Kruskal-Wallis test in swab or tissue specimens,respectively.The relative abundance of significant genus was brought into ordinal logistic regression model to draw receiver operating characteristic (ROC) curve.Area under the curve (AUC),specificity,sensitivity and akaike information criterion(AIC) were calculated for each model.The 10-fold crossvalidation was used as the internal validation method,and normalized mean square error (NMSE) was calculated.In this study,a conventional level of significance (P<0.05) was used for rejecting the null hypothesis.
Results
Baseline characteristics of participants
According to the pathological diagnosis,a total of 236 cases enrolled in this study were assigned to the normal group(70 cases),esophagitis group (all 70 cases were with mild esophagitis),LGIN group (62 mild dysplasia cases and 8 moderate dysplasia cases),HGIN group (9 severe dysplasia cases and 10 carcinomain situcases) and ESCC group (7 cases).The average age was significant different among groups (P<0.01) and there was an increasing tendency with the progression of ESCC (Mean agenormal=51.89 years,Mean ageESCC=59.20 years).Education background was statistical different among groups (P<0.05).Different from more normal and esophagitis participants with secondary or above education,participants with LGIN,HGIN and ESCC were mostly in primary or below education.There were no significant differences among groups in other personal information,life style and dietary habits (Table 1).
Microbial diversity between two esophageal specimens
This study examined the microbial diversity of paired esophageal biopsy and swab specimens to explore the collection methods of the two types of esophageal samples.There was no statistical difference of microbial diversity between swab and biopsy specimens (OTUs,P=0.71;Shannon index,P=0.49) (Table 2).Similarly,by comparing paired specimens of participants in each group respectively,no statistical difference was shown both for OTUs and Shannon index.However,on the whole,the microbial diversity was higher in swab samples than in biopsy samples,including OTUs (Biopsy=116.00,Swab=115.50)and Shannon index (Biopsy=4.39,Swab=4.34) (Table 2).About β diversity,significant clustering was detected for the weighted UniFrac distance (P<0.05,Figure 1A) and unweighted UniFrac distance (P<0.05,Figure 1B).
Microbial diversity of five participant groups
On the basis of comparison between two sampling methods,this study examined the microbial diversity by five participant groups to explore the dynamic changing during progression of ESCC.Within all biopsy samples,no statistical difference was detected among five groups(OTUs,P=0.13;Shannon index,P=0.09) but there was a decreasing tendency of OTUs from normal to ESCC.Within all swab samples,there was significant difference of OTUs (P<0.01) among groups but not Shannon index(P=0.26) (Figure 2).And partly similar to OTUs of biopsy samples,there was an inconspicuous decreasing of observed OTUs in swab samples from normal to HGIN and an inverse increasing tendency to ESCC.About β diversity,significant clustering was detected for the weighted UniFrac distance (P<0.05,Figure 1C) and unweighted UniFrac distance (P<0.05,Figure 1D).
Characteristic phylum and genus with progression of ESCC
The top 5 phyla in biopsy and swab specimens includeFirmicutes,Proteobacteria,Bacteroidetes,ActinobacteriaandFusobacteria(Figure 3).No significant statistical difference was discovered for any dominant phylum among groups in both collections.In swab specimens,there was statistical difference ofFirmicutesandFusobacteriain ESCC comparing with the normal group.And in both biopsy and swab specimens,there was a decreasing tendency ofFirmicuteswith the progression of ESCC,while the changing tendency ofFusobacteriawas opposite.At genus level,the top 10 genera in biopsy and swab specimens includeStreptococcus,Prevotella,Veillonella,Actinobacillus,Haemophilus,Neisseria,Alloprevotella,Rothia,GemellaandPorphyromonas(Figure 3).In biopsy samples,Neisseriawas significant different among groups with an increasing tendency from normal to ESCC.Besides,Streptococcus(Normal=0.1951, ESCC=0.0878, P<0.05),Haemophilus(Normal=0.0541,LGIN=0.0675,P<0.05) andPorphyromonas(Normal=0.0090, Esophagitis=0.0093,P<0.05) were meaningful in biopsy specimens.In swab samples,StreptococcusandNeisseriawere significant different among groups,with a decreasing and increasing tendency from normal to ESCC,respectively.And there was statistical difference ofHaemophilusin swab(Normal=0.0487,ESCC=0.0881,P<0.05) (Figure 4).The relative abundance ofPrevotella(P<0.05),Neisseria(P<0.05),Rothia(P<0.01) andPorphyromonas(P<0.05) was higher in swab than in tissues of normal group.In LGIN group,Prevotella(P<0.001),Veillonella(P<0.05) andRothia(P<0.05) were all higher in swab than in tissues butGemella(P<0.05) was opposite.
Microbial prediction model of ESCC and its precancerous lesions
According to the results of characteristic genera with theprogression of ESCC in two specimens,Streptococcus,Neisseria,HaemophilusandPorphyromonaswere taken into the microbial prediction model of ESCC and its precancerous lesions.To explore a better one,this study carried out all combinations of the above four characteristic genera.Only taken one genus into model,the AUC from high to low was as follows:0.732 (Neisseria),0.712(Haemophilus), 0.697 (Streptococcus) and 0.696(Porphyromonas).About 6 combinations of any two genera,the AUC ofNeisseriaandStreptococcuswas the best(0.738),and the AUC ofNeisseriaandPorphyromonasorHaemophiluswas 0.733 and 0.732,respectively.BringNeisseria,StreptococcusandPorphyromonasorHaemophilusinto model,the AUC was the same (0.738) and higher than the other two combinations of three genera.Taken all genera into model,the AUC was 0.738.Of all the combinations,the AUC of four combinations was the highest and same,which was 0.738 (Figure 5).Synthesizing several evaluation indexes of the four models,including sensitivity, specificity and AIC, the combination ofStreptococcusandNeisseriawas a more appropriate microbial prediction model of ESCC and its precancerous lesions.And we utilized the 10-fold cross-validation as the internal validation method,the NMSE was 1.575.
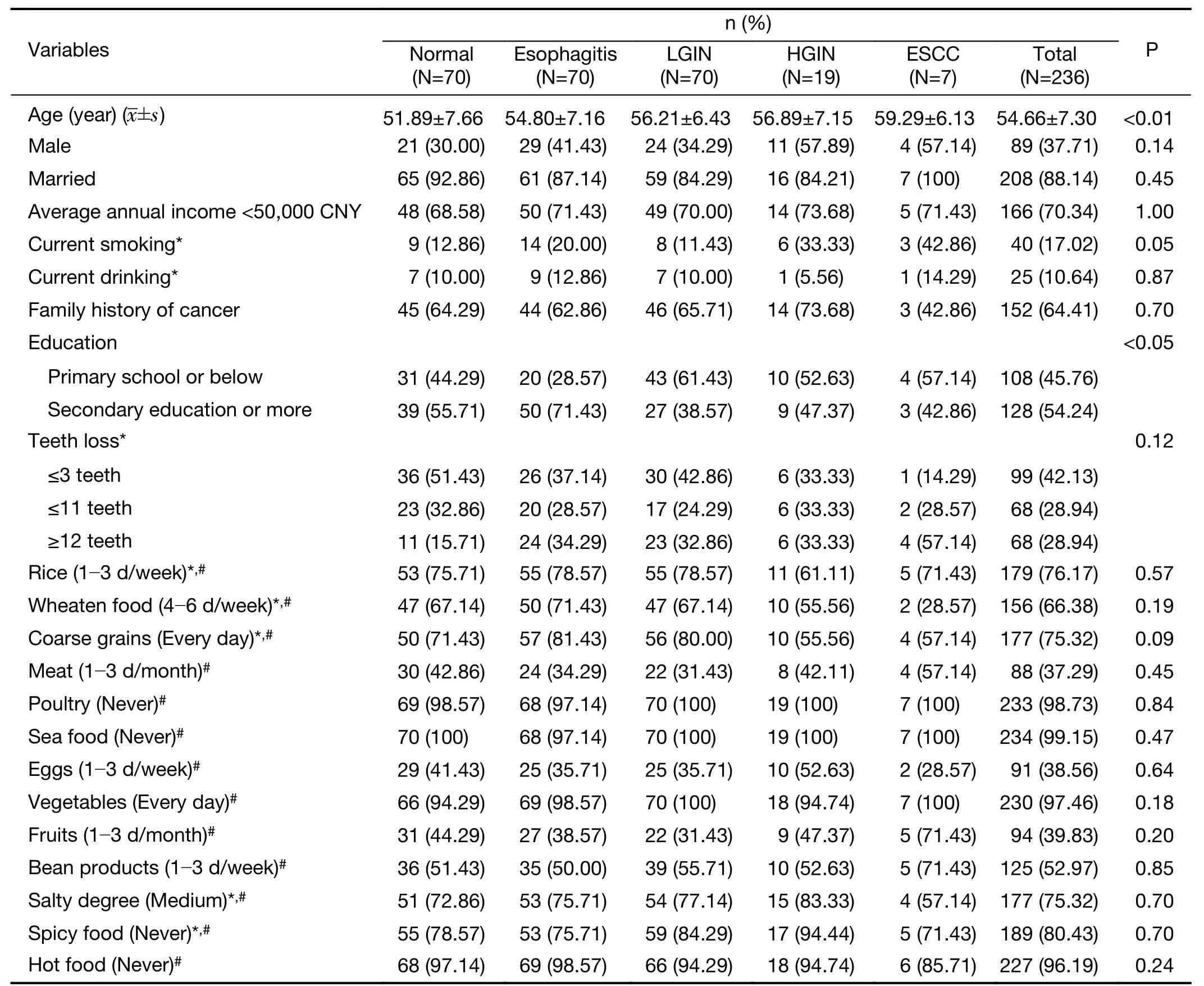
Table 1 Baseline characteristics of participants
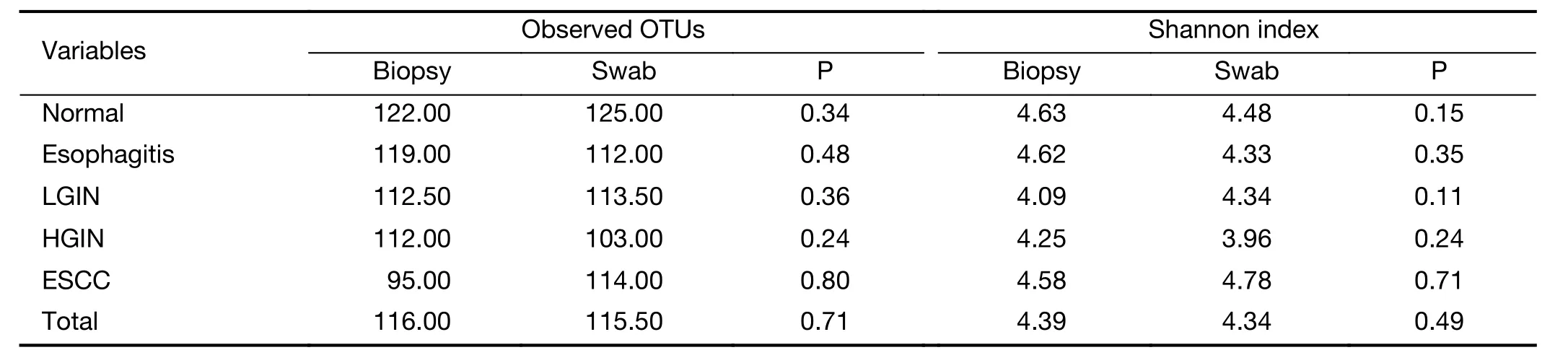
Table 2 α diversity between esophageal biopsy and swab specimen
Discussion
The esophageal microbiota includes various passenger microbes transiting from the oral cavity to the stomach and resident bacteria closely associated with the esophageal epithelium.In this study,we have observed and compared the esophageal microbiota that was closely associated with the esophageal mucosa under distinct pathological conditions to investigate the role of esophageal microbiota in the development of ESCC.
Combining endoscopic iodine staining and indicative biopsy is the gold diagnosis standard for ESCC and squamous dysplasia in China.Given the invasion of endoscopic biopsy and emerging of cytology,to enrich and verify previous studies,we flexibly compared the microbial composition of esophageal swab and biopsy specimens from normal,esophagitis,LGIN,HGIN to ESCC.In this study,α diversity was not significantly different between two specimens in any group,while mucosal swab specimens had a better performance than biopsy,which was similar to the findings conducted by Liu and Gall (11,23).Gallet al.(23)found a significant higher diversity in swab specimens compared with biopsies in esophagus.Another meaningful study (24) indicated that mucosal biopsies should remain the gold standard for microflora analysis in the esophagus,however,the swab specimens were from uvula and endoscope.Similar study in intestinal tract (12) revealed both swab and biopsy collection methods provided indifferent assessments of the microbial community and recommended mucosal brushing.Additionally,no significant difference was shown in microbial community in esophageal biopsy and swab specimens.Taken findings from this study and literatures together,both collection methods in microbial studies have pros and cons,which means appropriate collection methods are needed to depend on situation.
High-relative abundance genera includingStreptococcus,Prevotella,Veillonella,Actinobacillus,Haemophilus,Neisseria,Alloprevotella,Rothia,GemellaandPorphyromonaswere commonly seen in all samples from all participant groups,which was not exactly similar to findings of Dong and Pei (13,25).About the stable dominant genera community,there were various changes in relative abundance not only between two specimens but among participant groups.Comparing with healthy control,individuals with esophageal dysplasia had a significantly lower number of genera in esophagus in Yu’s finding (26).However,due to unbalanced sample size in this study,the decreasing tendency was inconspicuous and insignificant comparing with that of Yu (26).Yanget al.(27) proposed there was a shift from gram-positive microbiota to gram-negative microbiota in the microenvironment of the histologically abnormal distal esophagus.OnlyStreptococcusandGemellawere grampositive microorganisms among top 10 genera in our study.AndGemellawas one of the high-relative abundance genera in UGI (28) but not mentioned in Dong’s finding (25).
All esophageal samples were taken from middle or lower esophagus in this study,which had no influence on the microbial preference (25).Both in biopsy and swab specimens,there was no difference in α diversity between normal control and the other groups but relative abundance was slightly fluctuating.A previous study about the UGI microbiota has shown the lower microbial richness was significantly associated with the presence of esophageal squamous dysplasia (26).Shaoet al.(8) found there was no significant difference in α diversity for ESCC tumor and non-tumor tissues.Through various comparisons,the differences inStreptococcus,Neisseria,HaemophilusandPorphyromonaswere detected in swab or tissue specimens.Streptococcus,gram-positive genus,was one of the dominant genera in health esophagus,while the other three gram-negative genera were more likely to be dominant in esophagitis and Barrett’s esophagus (13,27).Among four significant genera in this paper,interestingly,onlyStreptococcushad a decreasing tendency from normal to ESCC,while the other genera had an increasing tendency.The dynamic changing ofStreptococcusin this study was a comprehensive complement of another microbial characteristic study that the relative abundance ofStreptococcusdecreased with more advanced ESCC (8).And this study firstly found there was a significant increasing tendency ofHaemophilusandNeisseriawith the progression of ESCC.Previously,HaemophilusorNeisseriaenrichening in eosinophilic esophagitis (EoE) has been found in findings conducted by Harris and Benitez (29,30).
Taken several evaluation indexes into consideration,combiningStreptococcusandNeisseriainto microbial prediction model of ESCC and its precancerous lesions had an ideal outcome.Interesting,genusNeisseriaof the phylumProteobacteriawas more abundant in the oral cavity than in esophagus,which was opposite to genusStreptococcusof the phylumFirmicutes(25).However,notNeisseriabutPrevotellahas been discussed in limited bacterial model studies.A prognosis study of ESCC revealed that combinedStreptococcusandPrevotellaabundance was an independent prognosis biomarker in ESCC patients (31),which did not discuss about squamous dysplasia.Analysis of esophageal brush samples from EAC participants has shown distinct community cluster ofStreptococcusandPrevotella(32).A study (8) about gastric cardia adenocarcinoma has shown the relative abundance ofStreptococcusdecreased andPrevotellaincreased with a more advanced tumor stage even these findings were not statistically significant.Prevotellawas associated with carbohydrates and simple sugars (33).Nobelet al.(34)found low fiber intake was associated with increased relative abundance of several gram-negative bacteria,includingPrevotella,Neisseriaand so on.All participants in this study were from the same county and had a similar dietary pattern.And as the progression of ESCC,the diet was easier to be affected.Boldly postulating,participants with higher disease degree,dietary intake may be reduced,and the relative abundance ofNeisseriaassociated with low fiber intake may be higher,which needs to be tested and verified in the future.And this study is an attempt to discuss the microbial model about the progression of ESCC.The AUC ofStreptococcusandNeisseriain this study was 0.738 which was lower than that in the study of Hsiehet al.(10),but not comparable.Hsiehet al.(10)figured out increased abundance ofClostridiumandFusobacterium,which exhibited a diagnostic ability for gastric cancer,and the AUC was 0.875.And unlikeStreptococcus,PrevotellaandFusobacteria,to our knowledge,this is one of the few highlighting the potential prediction ofNeisseria,which enriches previous findings and provides new direction for future studies (10,31,35).
Due to difficulty in sampling,few studies have explored the signatures within esophageal microbiome with the progression of ESCC.And this is an attempt to dynamically depict the microbial composition covering the whole progression of ESCC.However,there are still several limitations in this study.First,all participants in this study were from the natural population in Linzhou.Due to the low detection rate of precancerous lesions and cancer in natural population and limited screening time,there was an unbalanced but reasonable sample size for each group from normal to ESCC.Second,all swab and biopsy samples were collected by the same technological process that swab specimens were preserved in cell preserving fluid.This study did not extract DNA from the cell preserving fluid to further exclude the potential influence.Third,interaction among diet,microbiota and health is complicated and unclear (36).Age may contribute to but not fully explain the different esophageal microbial community types.For example,age is positively correlated with the relative abundance ofStreptococcus(32).In this study,all individuals were with a similar dietary pattern,but there was an aging tendency in different groups as the progression of ESCC,which was hard to diminish or exclude this interaction.
Conclusions
Returning to the two points posed at the beginning of this study.Esophageal swab specimens from participants with different histopathologic diagnosis have similar microbial signatures as corresponding esophageal biopsy,which provides sampling suggestion for future microbial studies.With the progression of ESCC,dynamic changing of highrelative abundance genera was informative and complicated.StreptococcusandNeisseriamay predict ESCC and its precancerous lesions but it is hard to ensure the causality between microbiota and ESCC progression.In the future,more attention needs to be paid to lowrelative abundance genera to enrich and perfect conclusions found by this study.And more large-scale case-control studies and prospective cohort studies are needed to further explore the dynamic bacterial changing in ESCC progression.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No.81974493) and the Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences (No.2016-I2M-3-001).
Footnote
Conflicts of Interest:The authors have no conflicts of interest to declare.
杂志排行

Chinese Journal of Cancer Research的其它文章
- Separate lateral parametrial lymph node dissection improves detection rate of parametrial lymph node metastasis in early-stage cervical cancer:10-year clinical evaluation in a single center in China
- Hyperthermic intraperitoneal chemotherapy for gastric cancer with peritoneal metastasis:A multicenter propensity scorematched cohort study
- Development and validation of prognostic nomogram based on log odds of positive lymph nodes for patients with gastric signet ring cell carcinoma
- Prognostic factors affecting long-term outcomes in patients with brain metastasis from esophageal carcinoma
- Oral microbiome and risk of malignant esophageal lesions in a high-risk area of China:A nested case-control study
- Current epidemiology of pancreatic cancer:Challenges and opportunities